
10. Genetic liver diseases
10.1 Haemochromatosis
Claus Niederau
Abstract
Hereditary haemochromatosis may be classified into five subtypes. The most frequent type 1 is an autosomal recessive trait affecting the HFE gene on chromosome 6. The homozygous C282Y mutation accounts for more than 90% of phenotypes in Caucasian populations and leads to an increase in intestinal iron absorption with a long-term risk of iron overload and organ damage including liver cirrhosis and diabetes mellitus. Early diagnosis in a non-cirrhotic stage and iron removal by phlebotomies are associated with a normal life expectancy.
This review will focus on type 1 HFE haemochromatosis. Other genetic types affect hemojuvelin, hepcidin, transferrin receptor 2, ferroportin 1, and bone morphogenetic protein 6.
Secondary haemochromatosis is usually caused by multiple blood transfusions in haemolytic anaemias such as thalassaemia, sickle cell anaemia, and myelodysplasia. Here, iron may accumulate faster than in genetic haemochromatosis leading to cardiomyopathy and liver cirrhosis. Therapy in secondary haemochromatosis consists of iron chelators because phlebotomies cannot be done due to anaemia.
Definition and classification of iron overload diseases
Hereditary haemochromatosis may be classified into five subtypes (Table 1). Type 1 is the well-known form of iron overload due to an autosomal recessive genetic metabolic malfunction; the homozygous C282Y mutation of the HFE gene on chromosome 6 accounts for more than 90% of clinical phenotypes in populations of Caucasian origin (Feder 1996). This mutation leads to an inadequately high intestinal iron absorption that after decades may cause iron overload and damage to various organs (Figure 1). Types 2a and 2b of genetic haemochromatosis are juvenile forms of iron overload that lead to a severe outcome prior to age 30, with cardiomyopathy and hypogonadism. The corresponding mutations are located in the hemojuvelin and hepcidin genes, respectively (Roetto 1999). Type 3 has mainly been described in Italian families and refers to a mutation in the transferrin receptor 2 gene (Girelli 2002). Clinical consequences of type 3 haemochromatosis are similar to type 1. Types 2 and 3 are autosomal recessive traits. The mutations of the autosomal dominant type 4 haemochromatosis are located in the gene coding for the basolateral iron transporter ferroportin 1 (Njajou 2001). In contrast to the other types, iron is accumulated in type 4 mainly in macrophages; ferritin values are markedly elevated although transferrin saturation is only slightly higher. Mutations in the gene of the bone morphogenetic protein 6 (BMP6) can also lead to a clinically often mild hepatic iron overload and may be defined as type 5 (Andriopoulos 2009, Meynard 2009, Daher 206). Neonatal haemochromatosis is characterised by severe fatal liver disease due to iron overload. Gestational alloimmune liver disease (GALD) has been established as the cause of fetal liver injury in the majority of cases with neonatal haemochromatosis (Feldman 2013). Probably, neonatal haemochromatosis is not a genetic disease (Feldman 2013, Kelly 2001, Hardy 1990). Further, non-classified rare iron overload syndromes include iron overload in Bantu Africans (Senba 1989) and iron overload in acaeruloplasminaemia (Doyle 2015).
Secondary haemochromatosis is usually caused by multiple blood transfusions in hemolytic anaemias such as thalassaemia, sickle cell anaemia and myelodysplasia syndrome. Iron first accumulates in RES macrophages and is later transferred to parenchymal cells. With frequent blood transfusions, iron may accumulate faster than with genetic haemochromatosis; iron overload often leads to severe cardiomyopathy and liver cirrhosis, limiting effective prognosis. Therapy consists of iron chelators because phlebotomies cannot be done due to the underlying anaemia. This review will focus on type 1 HFE haemochromatosis, the most prevalent genetic form in Germany. Most consequences of iron overload are similar, whatever the cause. Thus, the pathophysiology of tissue and organ damage by iron excess is discussed in detail only for HFE haemochromatosis.
 Figure 1. Scheme of natural history of type 1 genetic haemochromatosis (HFE)
Table 1. Classification of haemochromatosis
Figure 1. Scheme of natural history of type 1 genetic haemochromatosis (HFE)
Table 1. Classification of haemochromatosis
| I) Genetic haemochromatosis | ||||
| Types | Gene defect on | Affected gene | Inheritance | High prevalence |
| Type 2a | Chromosome 1 | Hemojuvelin | Autosomal recessive | Juvenile form |
| Type 2b | Chromosome 19 | Hepcidin | Autosomal recessive | Juvenile form |
| Type 3 | Chromosome 7 | Transferrin receptor 2 | Autosomal recessive | Italy |
| Type 4 | Chromosome 2 | Ferroportin 1 | Autosomal dominant | Italy |
| Type 5 | Chromosome 6 | BMP6 | Autosomal dominant | Panethnic? |
| Others | Unknown | Unknown | Unknown | Of non-Caucasian origin |
| II) Neonatal haemochromatosis, in most cases caused by non-genetic gestational alloimmune liver disease (GALD) | ||||
| III) Secondary haemochromatosis | ||||
|
a) Chronic anaemias (thalassaemia, sickle cell disease, MDS, other rare hemolytic anaemias) b) Multiple blood transfusions in general c) Long-term oral intake of high amounts of iron (diet-related or intravenous) |
||||
| IV) Dysmetabolic Iron Overload Syndrome (DIOS) usually associated with metabolic syndrome and fatty liver disease | ||||
| V) Further, non-classified iron overload syndromes | ||||
|
a) iron overload in Bantu Africans b) iron overload in aceruloplasminaemia |
||||
Type 1 HFE haemochromatosis
History
The association between liver cirrhosis, pigment deposits in the liver, and diabetes mellitus was recognised over a century ago (Trosseau 1865, Troisier 1871, Hanot and Schachmann 1886). The term haemochromatosis was first introduced in the 19th century (Recklinghausen 1889) but was not generally accepted until used as the title of a classic monograph (Sheldon 1935). The controversy over whether haemochromatosis was merely a form of alcoholic liver cirrhosis (MacDonald 1960) or a genetic error of iron metabolism (Sheldon 1935, Crosby 1966) lasted almost a century until the association between special HLA haplotypes and haemochromatosis which recognised the genetic nature of the disease was described (Simon 1975). The mode of inheritance was identified as an autosomal recessive disorder (Simon 1977). Finally, the major mutation on the HFE gene associated with clinical manifestations was identified (Feder 1996).
Epidemiology
Type 1 haemochromatosis is probably the most prevalent genetic metabolic error in Caucasian populations (Adams 2005). The prevalence of C282Y homozygotes is approximately 0.5% in central Europe and in the Caucasian population of North America; the prevalence of C282Y and H63D heterozygotes approaches 40% in similar populations (Adams 2005). Phenotypic expression also depends on several non-genetic factors such the amount of dietary iron and blood loss (Figure 2). For example, due to menses, females develop clinical consequences of iron overload 5–8 times less frequently and 10–20 years later than males. It is now widely accepted that not all C282Y homozygous men will develop the full clinical manifestation of haemochromatosis. It also remains unclear how many men will show clinical disease during their lifetime and what factors determine that phenotype.
As mentioned previously, the homozygous C282Y mutation accounts for more than 90% of the clinical phenotype in Caucasian populations (Feder 1996, Adams 2005) (Table 2). A point mutation at H63D is also frequently identified in the HFE gene as well as other less frequent mutations. None of these gene alterations or polymorphisms, found in up to 40% of Caucasians, correlates with the phenotype. A subject with a C282Y variation on one allele and a H63D variation on the other is called a “compound heterozygote” (Table 2). Only a small percentage of such compound heterozygotes are at risk for clinical consequences of iron overload (Gallego 2015). A recent meta-analysis showed a positive association between compound heterozygosity for C282Y/H63D and the risk of NAFLD and HCC, but not liver cirrhosis (Ye et al. 2016). Patients who are compound heterozygous for C282Y/H63D or homozygous for H63D with confirmed iron overload should be investigated for other causes of iron overload and may be treated with phlebotomy on an individual basis (EASL 2022); the benefit of phlebotomy is however largely unclear in the latter patients. C282Y and H63D heterozygotes are at no risk of iron overload (Table 2). In non-Caucasian populations other genes may be involved in causing iron overload.
Aetiology and pathogenesis
Intestinal iron absorption and iron losses are finely balanced under physiological conditions. Approximately 10% of the total daily intake of iron (10–20 mg) is absorbed by the small intestine (1–2 mg). However, subjects with the homozygous C282Y mutation may absorb up to 20% of iron intake; i.e., up to 2–4 mg/day. Thus, homozygotes have an excessive iron intake of approximately 1 mg/day. It may therefore take several decades until iron stores approach 10 g, above which organ damage is considered to start. Many patients at the clinical end stage of haemochromatosis, including liver cirrhosis and diabetes mellitus, have total body iron stores of 20–30 g. Intestinal iron absorption is downregulated when iron stores increase in these patients, as it is in patients with genetic haemochromatosis. This downregulation, however, occurs on an increased level when compared to subjects without the HFE gene mutation. Correspondingly, intestinal iron absorption is massively increased in patients with haemochromatosis when iron stores have been depleted by phlebotomy. It is important to continue phlebotomies after iron depletion in order to prevent reaccumulation (see Table 4). These regulatory processes however do not explain how HFE gene mutations cause the increase in intestinal iron absorption since the HFE gene product is neither an iron transporter nor an iron reductase or oxidase. However, carriers and regulators of cellular iron uptake and release been identified (Pietrangelo 2002, Fleming 2002, Townsend 2002, Fletcher 2002).
Some of these carriers also interact with the HFE gene product in the regulation of intestinal iron absorption (Pietrangelo 2002, Fleming 2002, Townsend 2002, Fletcher 2002) and the Nramp2 protein is the luminal iron carrier. Luminal iron reductase has also been identified as the Dcytb protein (duodenal cytochrome B) (Pietrangelo 2002, Fleming 2002, Townsend 2002, Fletcher 2002). The basolateral iron transporter ferroportin 1 (also named Ireg1 or MTP1) has also been identified (Donovan 2000, Abboud 2000) as well as the basolateral iron oxidase haephestin (Vulpe 1999). Mutations in some of these proteins are responsible for the rarer types 2–4 of genetic haemochromatosis, although none of these genes is altered in type 1 haemochromatosis. Two other proteins have been shown to act as important iron regulating proteins, transferrin receptor 2 and hepcidin (Pietrangelo 2002, Fletcher 2002, Fleming 2005). Mutations in the transferrin receptor 2 gene may lead to the rare type 3 haemochromatosis, and mutations in the ferroportin 1 gene to type 4 haemochromatosis. More recent studies also indicate that hepcidin may be the most important regulator of iron metabolism, involved in iron deficiency and overload. Hepcidin has been shown to downregulate the basolateral iron carrier ferroportin. It has also been demonstrated that hepcidin itself is upregulated by HFE. Thus, an HFE mutation may reduce the upregulation of hepcidin that then does not downregulate ferroportin; the corresponding increase in ferroportin expression finally causes the increase in intestinal iron uptake (DeDomenico 2007). The BMP-SMAD pathway in addition plays an important role in regulating hepcidin to control systemic iron homeostasis (Feldmann 2013). There may be further interactions between HFE, transferrin receptor 2, Nramp2, Dcytb, ferroportin, haephestin, BMP6, and hepcidin.
The penetrance of genetic HFE haemochromatosis depends on various non-genetic factors and genetic co-factors such as HCP1, GNPAT, PCSK7, TM6SF2, PNPLA3 and MBOAT7 (Niederau 2018) (Figure 2).
The Dysmetabolic Iron Overload Syndrome (DIOS) is usually associated with the metabolic syndrome and fatty liver disease; it is further characterised by an increase in serum ferritin with normal/slightly elevated transferrin saturation and only slightly elevated liver iron (Datz 2017, Deugnier 2017, Fernandez 2022). The increase in liver iron may accelerate fibrosis in patients with DIOS. The pathogenetic mechanisms linking metabolic abnormalities, insulin resistance, and fatty liver disease to iron accumulation are still ill defined. There is as yet no evidence that iron removal by phlebotomies has a benefit for the patient with DIOS.
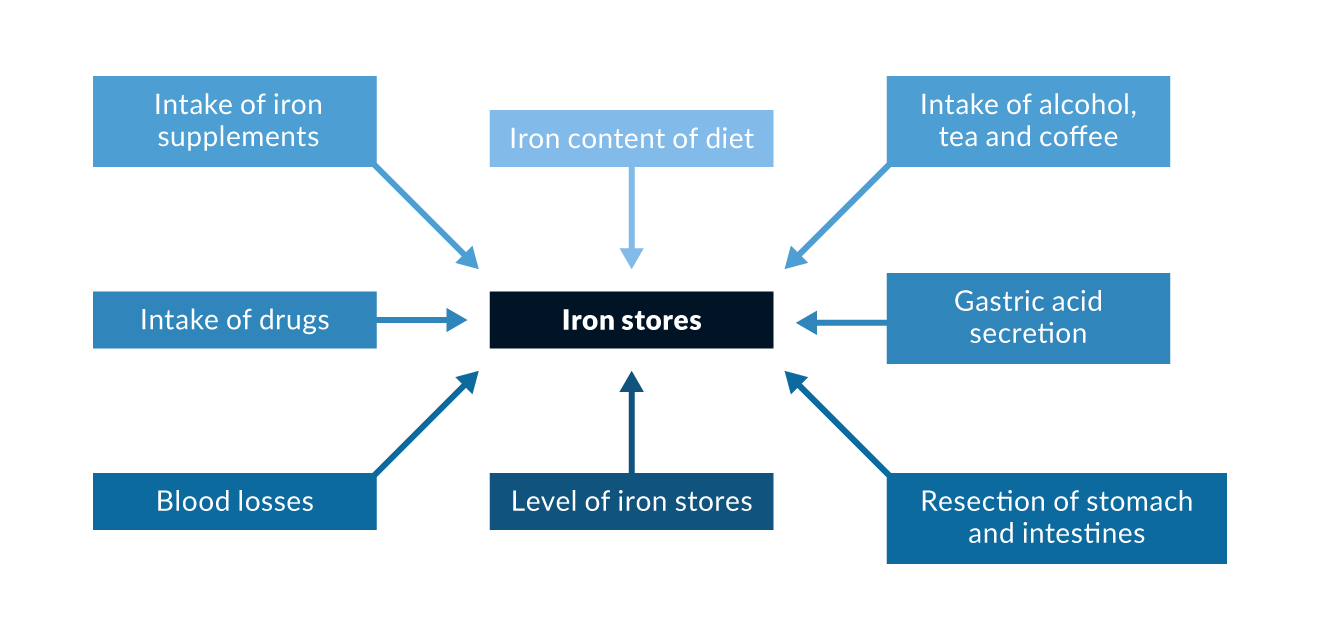 Figure 2. Non-genetic factors and genetic co-factors that may influence iron absorption
Table 2. Genotype/phenotype correlation in haemochromatosis
Figure 2. Non-genetic factors and genetic co-factors that may influence iron absorption
Table 2. Genotype/phenotype correlation in haemochromatosis
| Mutations/polymorphisms | Prevalence in Caucasian populations | Risk of advanced clinical phenotype |
| C282Y/C282Y | 85–95% | low if ferritin is <1000 ng/mL |
| H63D/C282Y | 3–8% | very low |
| C282Y/wild type | - | none |
| H63D/wild type | - | none |
| Others | 1 % | unknown |
Diagnosis
Laboratory tests. Any increase in serum iron should start with the exclusion of haemochromatosis so as not to overlook early disease. Normal serum iron, however, does not exclude haemochromatosis, and increased serum iron often occurs in the absence of haemochromatosis. Serum iron values are highly variable and should not be used either for diagnosis or for screening of haemochromatosis. The determination of transferrin saturation is a better indicator of iron overload than serum iron. The increase in transferrin saturation usually precedes the ferritin increase (Figure 1). Transferrin saturation is more sensitive and specific for detection of haemochromatosis when compared to serum ferritin. For screening, a threshold of 50% for transferrin saturation in men and 45% for women may be optimal under fasting conditions (EASL 2022). Ferritin on the other hand is a good indicator of largely increased iron stores and reliably indicates iron deficiency. It has less value for early detection of haemochromatosis.
In HFE haemochromatosis a slightly increased serum ferritin (300–500 ng/mL) is usually accompanied by transferrin saturations exceeding 80–90%. Unfortunately, serum ferritin is often increased also in the presence of infections and malignancies (regularly without a major increase in transferrin saturation), and thus has a low specificity for indicating haemochromatosis (Niederau 1998). Ferritin increases not due to genetic haemochromatosis are usually associated with normal or only slightly elevated transferrin saturation. Therefore, transferrin saturation should be measured in order to correctly interpret ferritin increases.
Liver biopsy, determination of liver iron concentration, and fibrosis assessment. Although simultaneous increases of both serum ferritin and transferrin saturation strongly indicate a risk for haemochromatosis, diagnosis needs to be confirmed by genetic testing or by liver biopsy with a determination of iron content in the liver. Hepatic iron concentration also increases with time in subjects with an HFE gene mutation. In order to obtain the “hepatic iron index”, divide the liver iron concentrations by the patient’s age. (Summers 1990). The semi-quantitative estimation of liver iron stores by the Berlin blue colour is less sensitive and specific than the chemical quantification of liver iron concentration. In case of a homozygous C282Y gene test, liver biopsy is not required for the diagnosis of genetic haemochromatosis (Figure 3).
There may, however, be other reasons to perform a liver biopsy in iron overload: (1) subjects with biochemical or clinical evidence of iron overload in the absence of the homozygous C282Y mutation should have a liver biopsy to substantiate iron overload and to look for hepatic comorbidities; (2) in C282Y homozygotes the risk for liver fibrosis and cirrhosis only increases at ferritin values >1000 ng/mL (Loreal 1992); in those patients liver biopsy may be useful because the presence of liver cirrhosis (and probably also of severe fibrosis) markedly increases subsequent risks of hepatocellular carcinoma (HCC) and thus warrants HCC surveillance. Patients with evident cirrhosis and those with a stiffness <6, 4 kPa do not need a liver biopsy (EASL 2022) (Figure 3). All patients with iron overload should have a non-invasive assessment of liver fibrose e.g. by elastography or FIB4 test (EASL 2022).
Deferoxamine testing and ferrokinetic measurements. Determination of urinary excretion of iron after administration of deferoxamine allows some estimation of total body iron stores. The deferoxamine test, however, often only shows pathological results when serum ferritin and transferrin saturation are markedly increased and does not allow diagnosis of early disease. Ferrokinetic measurements today are only done for scientific research or in difficult diagnostic situations.
Computed tomography (CT), magnetic resonance imaging (MRI), and biomagnetometry. CT density measurements of the liver allow a semi-quantitative estimation of iron concentration in the liver. This method however is associated with radiation and therefore not allowed in many countries where alternative methods are available. MRI, on the other hand, allows a reliable measurement of liver iron content, provided that special software is used and the equipment is calibrated for such measurement. In clinical practice most MRI do not fulfil these criteria. The recent EASL guidelines nevertheless state that iron overload should be assessed by MRI and not by liver biopsy any longer (EASL 2022). This recommendation may be difficult to observe in situations where no specific MRI is available and in situations where the costs for such MRI are not reimbursed by the health insurances.
Biomagnetometry allows the most accurate non-invasive measurement of liver iron concentration. However, this equipment is expensive and only allows measurement of iron concentration. Consequently, biomagnetometry is done only at a few centres worldwide and is primarily used for scientific studies and not in daily clinical practice. With the availability of reliable and inexpensive genetic testing, CT, MRI, and biomagnetometry are usually not needed for diagnosis of most patients with HFE haemochromatosis.
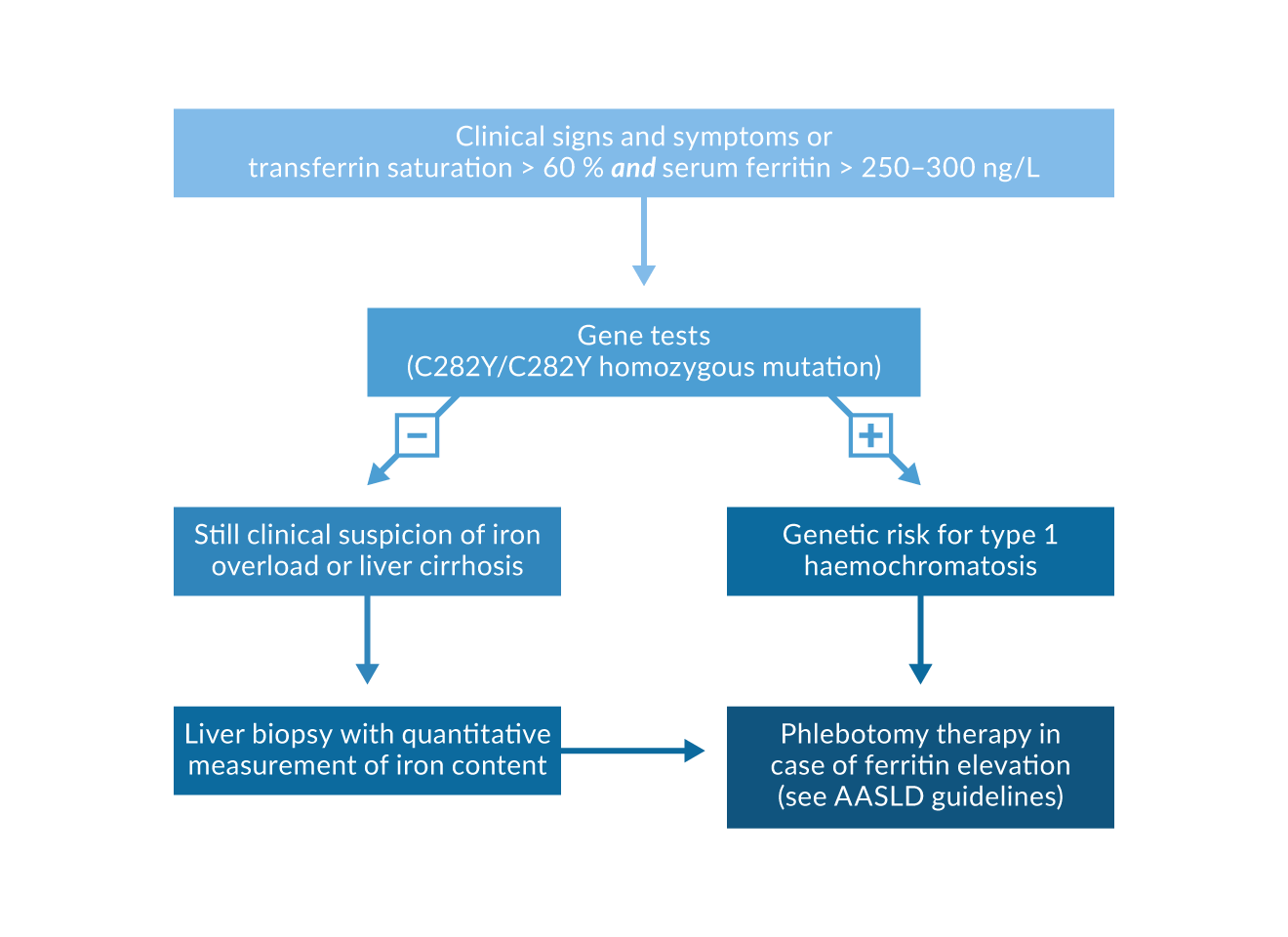 Figure 3. Diagnosis and treatment algorithm for type 1 haemochromatosis
Figure 3. Diagnosis and treatment algorithm for type 1 haemochromatosis
Genetic tests. As outlined previously, in Caucasian populations the homozygous C282Y mutation accounts for more than 90% of patients with the clinical phenotype of type 1 HFE haemochromatosis (Adams 2005, Erhardt 1999). Approximately 5% of patients with the clinical phenotype are C282Y/H63D compound heterozygotes; the prevalence of C282Y or H63D heterozygosity in patients with the clinical phenotype of haemochromatosis is considerably lower than in the general population. Thus, a subject who is heterozygous for C282Y or H63D per se has no risk of iron overload. In subjects homozygous for C282Y, both serum ferritin and transferrin saturation are frequently increased; however, only male subjects have an increased risk for liver disease when compared to subjects without HFE gene alterations in a recent large screening study (Adams 2005). It is unknown how many C282Y homozygotes will later develop clinical signs and symptoms due to iron overload. It is increasingly evident that only a minority of C282Y homozygotes progress to end stage iron overload with liver cirrhosis and diabetes mellitus. In subjects who are not C282Y homozygotes but have laboratory, histological or clinical evidence of iron overload, further genes may be analysed for mutations such as hemojuvelin, transferrin receptor 2, ferroportin 1 BMP6, and hepcidin.
Early diagnosis and screening
The prevalence of C282Y homozygotes is 0.5% in Caucasians (Adams 2005, Erhardt 1999). Clinical manifestations however are variable and depend on non-genetic factors such as dietary iron intake and blood loss. Until 1980, most patients with haemochromatosis were detected with late irreversible complications such as liver cirrhosis and diabetes mellitus. With a better understanding of the disease, the broad use of ferritin and transferrin saturation measurements and the availability of a reliable genetic test, diagnostic efforts have concentrated on the detection of early disease before liver cirrhosis and diabetes mellitus. Several studies have shown that iron removal by phlebotomy is associated with normal life expectancy in patients diagnosed early (Niederau 1985, Niederau 1996, Fargion 1992) (Figure 4). Several other studies have focused on screening procedures in order to diagnose more subjects with early disease (Edwards 1988). These studies include populations with special risks, family members, as well as the general population (Table 3) (Niederau 2002). It has been shown that an increasing number of patients are now diagnosed early and that this trend increases survival (Figure 5).
A large number of studies have shown that screening is useful for detection of asymptomatic C282Y homozygotes by using transferrin saturation and serum ferritin as well a genetic test for the C282Y mutation (Edwards 1988, Phatak 1998, Niederau 1998). A broad screening of the general population however is as yet not recommended by WHO and CDC mainly because it is unknown how many of the asymptomatic C282Y homozygotes will later develop clinical disease (see US Preventive Services Task Force 2007). The largest screening study analysed HFE gene mutations in almost 100, 000 subjects in North America. In Caucasians, C282Y homozygosity was found in 0.44%, a value similar to many previous studies in other populations with a similar background. Asian or Black people in contrast almost never have an HFE gene mutation (Adams 2005). Among the Caucasian C282Y homozygotes only males had a significant increase in liver disease when compared to subjects without an HFE gene variation (Adams 2005). Only further prospective follow-up studies will determine how many asymptomatic C282Y homozygotes will develop clinical consequences of iron overload.
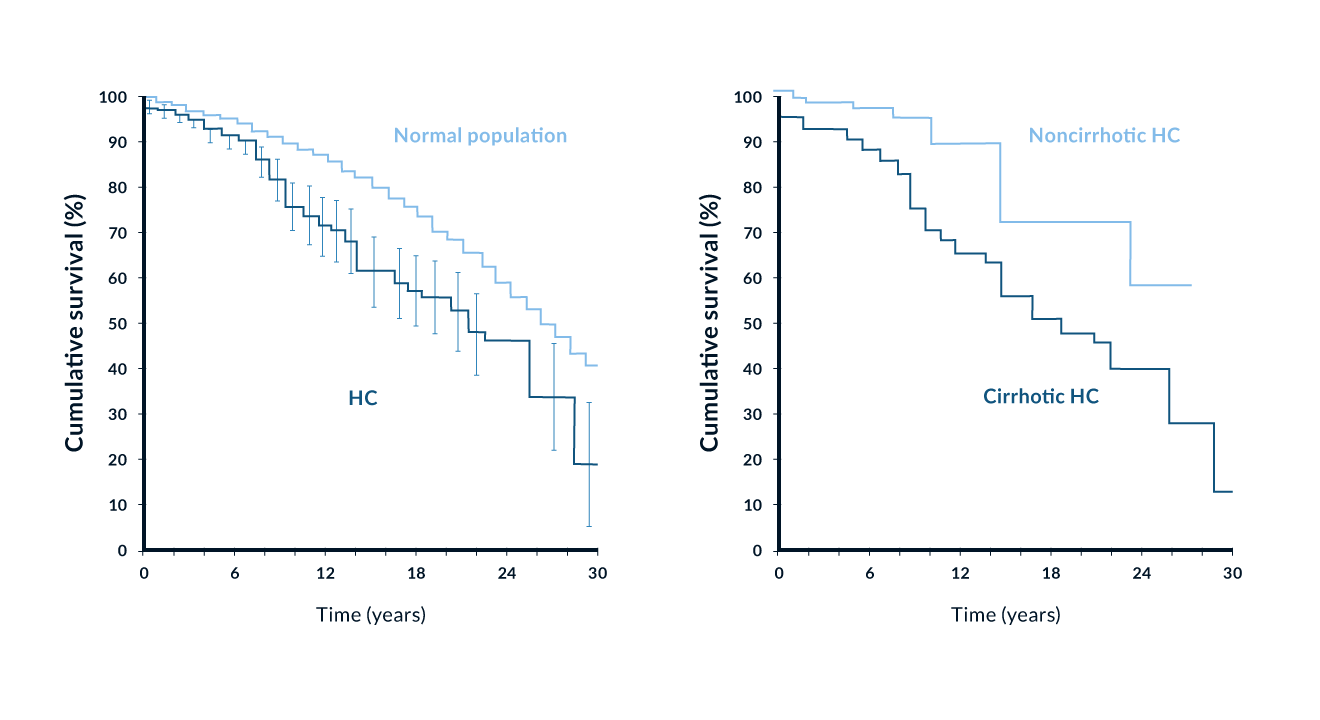 Figure 4. Survival of 251 patients with genetic haemochromatosis (with and without cirrhosis) in comparison with a matched general population. Modified from (Niederau 1996)
Table 3. Methods for early diagnosis of haemochromatosis
Figure 4. Survival of 251 patients with genetic haemochromatosis (with and without cirrhosis) in comparison with a matched general population. Modified from (Niederau 1996)
Table 3. Methods for early diagnosis of haemochromatosis
| 1. Screening in the general population not recommended |
| Screening of HFE gene alterations is not recommended in the general population because it remains unknown how many of the C282Y homozygotes will develop clinical manifestations. Such screening would be meaningful only in Caucasian populations. |
| 2. Family screening |
| Genetic testing can reliably determine who, among the first-degree relatives of a haemochromatotic patient, is a heterozygote or homozygote. Heterozygotes are healthy and do not need follow-up. C282Y homozygotes should be followed and treated by phlebotomy if ferritin increases >300 ng/mL in men and >200 ng/mL in women. |
| 3. Haemochromatosis should be excluded in patients with |
| newly diagnosed diabetes mellitus chronic liver disease of unknown aetiology elevation of iron, transferrin saturation or serum ferritin cardiomyopathy of unknown aetiology arthropathy of unknown aetiology loss of potency/libido and amenorrhea of unknown aetiology |
| 4. Every liver biopsy needs to be checked for iron deposits |
It is also unknown at which ferritin values phlebotomy treatment should be initiated in asymptomatic C282Y homozygotes (Table 4). The values recommended by the AASLD are based more on the judgment of experts than on solid data. The only solid data show that the risk for liver fibrosis and cirrhosis increases above the threshold of 1000 ng/mL for serum ferritin (Loreal 1996). The value of screening family members is obvious when a first-degree relative has clinical haemochromatosis. Such family screening is easy to do with the genetic test. Heterozygous family members are not at risk for haemochromatosis unless they have other risk factors.
The clinical phenotype of haemochromatosis is seen in 1–2% of patients with newly diagnosed diabetes mellitus and in 3–15% of patients with liver cirrhosis (Niederau 1999). These latter patients should be screened for iron overload although such screening obviously does not aim at a very early diagnosis. Nevertheless, cirrhotic and diabetic patients with haemochromatosis can benefit significantly from phlebotomy therapy. Little is known about the prevalence of haemochromatosis in patients with arthropathy or cardiomyopathy of unclear aetiology. Several smaller studies indicate that arthropathy may be a rather early clinical sign of iron overload, whereas cardiomyopathy usually occurs in late stage iron overload.
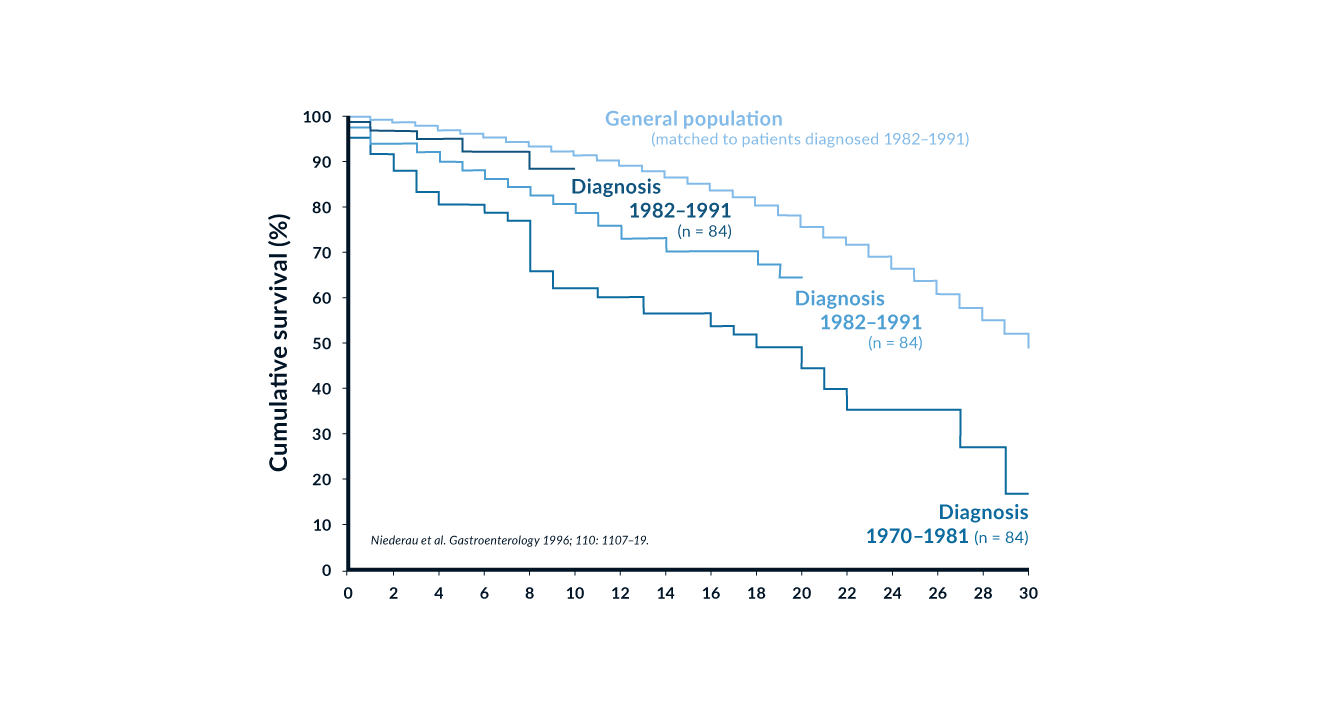 Figure 5. Cumulative survival in 251 patients with genetic haemochromatosis according to the time of diagnosis. Modified from Niederau 1996
Table 4. Iron overload therapy
Figure 5. Cumulative survival in 251 patients with genetic haemochromatosis according to the time of diagnosis. Modified from Niederau 1996
Table 4. Iron overload therapy
| 1. Phlebotomy |
| a) In symptomatic genetic haemochromatosis |
| Aims: complete iron depletion in 12-24 months; Treatment: 1-2 phlebotomies of 500 mL each week until serum ferritin is in the range of 50-100 ng/mL; Long-term therapy with 4-8 phlebotomies per year to keep ferritin between 50-100 ng/mL and thus prevent reaccumulation of iron |
| b) In asymptomatic C282Y homozygotes therapy should be initiated above these ferritin values: |
| Subjects <18 years >200 ng/mL Men >300 ng/mL Women (not pregnant) >200 ng/mL Women (pregnant) >500 ng/mL |
| 2. Therapy with iron chelators in secondary haemochromatosis and anaemia |
| Aims: removal of iron overload by increase of iron excretion in faeces and urine In case of further blood transfusions at high frequency at stabilisation of iron balance and reduction of further iron accumulation Treatment: until recently, 25-50 mg deferoxamine/kg as sc infusion for 10-12 h daily; today, deferoxamine is largely replaced by the oral chelator deferasirox – 20 mg/kg deferasirox once daily to prevent iron accumulation up to 800 mL erythrocytes concentrates/month Long-term treatment necessary Normalisation of ferritin and liver iron concentration often not possible |
| 3. Diet |
| Recommended: avoidance of food with very high iron content (e.g., liver) and iron-supplemented food; A further strict iron-depleted diet very difficult to adhere to and not recommended A single phlebotomy of 500 mL blood as effective for iron removal as a very rigid iron-restricted diet for a full year |
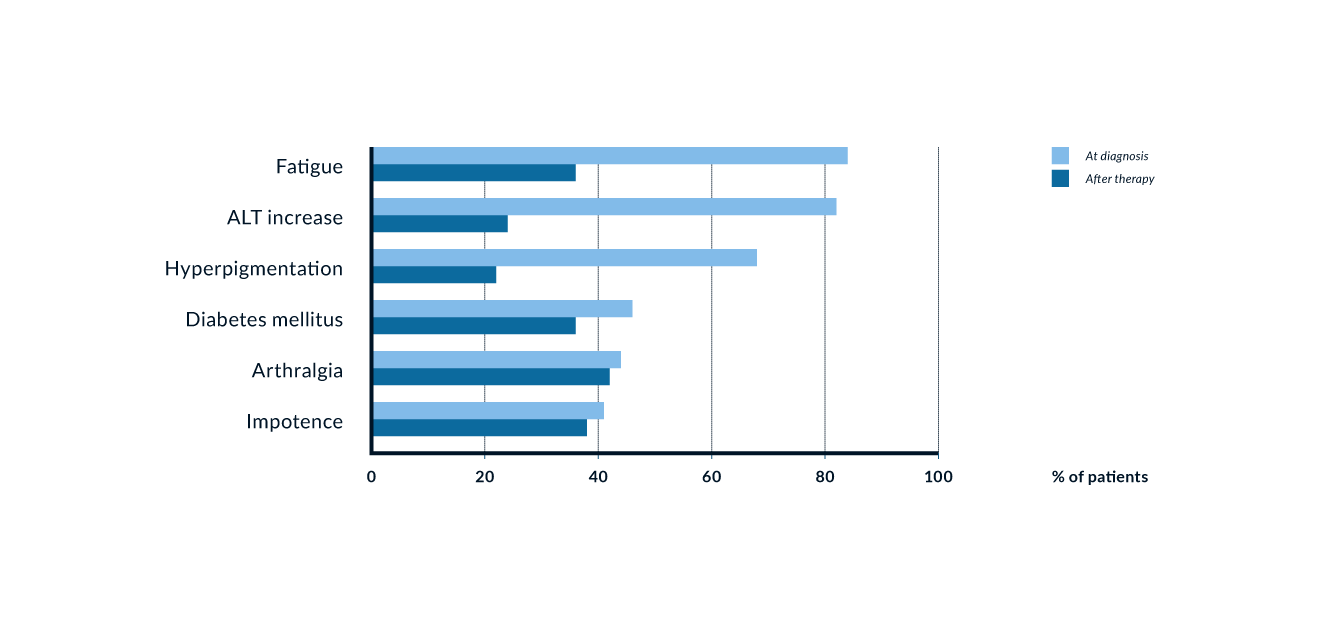 Figure 6. Signs and symptoms in 185 patients with genetic haemochromatosis prior to and after iron removal. Modified from Niederau 1996
Figure 6. Signs and symptoms in 185 patients with genetic haemochromatosis prior to and after iron removal. Modified from Niederau 1996
Complications of iron overload
Liver cirrhosis, diabetes mellitus and increased skin pigmentation are the classical trio of genetic haemochromatosis. Cardiomyopathy, cardiac arrhythmias and impotence are also typical complications of advanced iron overload. Arthropathy in contrast may be an early sign of haemochromatosis, which may help with diagnosis in the precirrhotic stage (Niederau 1996).
Liver disease. The liver is the organ that is affected by genetic iron overload most early and heavily. At early stages excess iron stores are mainly found in periportal parenchymal cells as ferritin and hemosiderin. When iron excess further increases, there is development of perilobular fibrosis and iron stores are also found in bile ducts and Kupffer cells. Septal fibrosis eventually progresses towards complete cirrhosis. The stage of fibrosis is closely associated with the degree of excess of iron. In many affected symptomatic patients with type 1 haemochromatosis there are some signs of liver disease at the time of diagnosis (Niederau 1985, Niederau 1996). Many nonspecific symptoms such as abdominal discomfort and fatigue may also be due to liver involvement. In asymptomatic patients diagnosed by a screening procedure, signs of liver disease are infrequent. Complications due to cirrhosis such as ascites, jaundice and portal hypertension are seen only rarely and only in cases of advanced severe iron overload (Niederau 1985, Niederau 1996). The risk for liver cirrhosis increases at ferritin values >1000 ng/mL (Loreal 1996). Similar to insulin-dependent diabetes, liver cirrhosis cannot be reversed by removal of iron (Niederau 1996). However, less advanced stages like hepatic fibrosis and abnormalities in liver enzymes and function respond well to iron removal (Niederau 1996) (Figure 5). Survival is significantly reduced in the presence of liver cirrhosis whereas patients diagnosed in the precirrhotic stage have a normal life expectancy when treated by phlebotomy (Niederau 1996) (Figure 4).
Association of haemochromatosis with other liver diseases. Some studies indicate that C282Y heterozygosity may aggravate the progression of concomitant liver diseases such as porphyria cutanea tarda, chronic hepatitis C, alcoholic hepatitis and non-alcoholic steatohepatitis (NASH). In these latter patients one might find slightly elevated liver iron concentrations and serum ferritin levels when they are C282Y heterozygotes (for review see Erhardt 2003). Most studies however have shown that these associations are of only minor importance in the clinical course of the disease. Phlebotomy has so far only been proven meaningful in porphyria cutanea tarda because it can ameliorate the cutaneous manifestations.
Liver carcinoma. Liver carcinoma develops in approximately 30% of patients with haemochromatosis and cirrhosis independent of iron depletion (Niederau 1996). The interval between complete iron depletion and reported diagnosis of liver cancer is approximately nine years in large cohorts in German patients (Niederau 1985, Niederau 1996). The risk of liver cancer is increased 100–200-fold in patients with haemochromatosis when compared to the general population (Figure 6). Among liver cancers there are hepatocellular carcinoma (HCC) as well as cholangiocellular carcinoma. Most liver cancers develop in patients with cirrhosis. Thus, cancer screening by ultrasound and potentially also by AFP (twice a year) is only recommended for patients with cirrhotis or severe fibrosis (EASL 2022). Patients who develop liver cancer usually have the largest amount of iron accumulation among various subgroups (Niederau 1996, Niederau 1999).
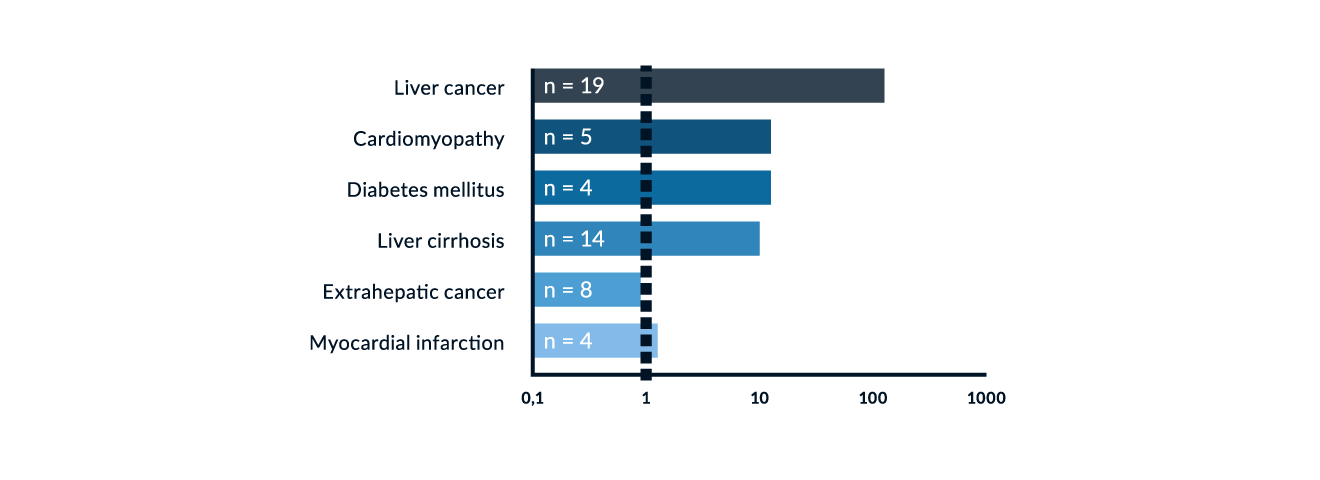 Figure 7. Relative mortality risk of 251 patients with genetic haemochromatosis in comparison to the general population. Modified from Niederau 1996
Figure 7. Relative mortality risk of 251 patients with genetic haemochromatosis in comparison to the general population. Modified from Niederau 1996
Diabetes mellitus. The prevalence of diabetes in hereditary haemochromatosis ranges from 20–50% (Niederau 1996, Adams 1991). The prevalence and stage of diabetes is related to the degree of iron deposition in the pancreas. Patients with diabetes have a twofold higher mobilisable iron content than non-diabetics (Yaouanq 1995). Investigations into the prevalence of unrecognised genetic haemochromatosis in diabetic patients show some variation in Europe vs. elsewhere; i.e., screening revealed a prevalence of 5–8 per 1000 unrecognised cases in Europe (Singh 1992) and 9.6 per 1000 in Australia (Phelps 1989). Diabetes mellitus and impaired glucose tolerance are frequent features in several chronic liver diseases (Creutzfeldt 1970, Blei 1982). This author’s study (Niederau 1984) showed hyperinsulinaemia and hence insulin resistance without impaired glucose tolerance in noncirrhotic haemochromatosis. The increase in circulating insulin concentrations is likely to be due to a decrease in diminished hepatic extraction of insulin. With the progression of iron overload and destruction of beta cells, insulin secretion becomes impaired (Dymock 1972, Bierens de Haan 1973). In end-stage haemochromatosis, insulin deficiency is associated with severe reduction in the mass of beta cells (Rahier 1987). Insulin resistance observed in early iron overload may be partially reversible after phlebotomy therapy (Niederau 1985, Niederau 1996) whereas insulin-dependent diabetes is irreversible (Niederau 1996). Survival is significantly reduced in patients with diabetes mellitus at diagnosis compared to patients without diabetes (Niederau 1996). Survival of non-diabetic patients is virtually identical to that of a matched normal population.
Heart disease. Cardiomyopathy and cardiac arrhythmias are specific complications of haemochromatosis caused by iron deposition in the heart (Buja and Roberts 1971, Short 1981). Clinical or electrocardiographic signs of heart disease can be found in 20–35% of patients with HFE haemochromatosis (Niederau 1985). Arrhythmias usually respond well to iron removal (Short 1981, Niederau 1996). In type 1 haemochromatosis cardiomyopathy is rare and usually associated with advanced iron overload and an older patient population. However, particularly in young patients who present with cardiac disease due to haemochromatosis, cardiomyopathy is a frequent cause of death (Finch 1966, Short 1981). It has also become clear that young patients with severe cardiomyopathy may be affected by juvenile type 2 haemochromatosis; these patients may show severe iron overload, hypogonadism, cardiomyopathy, liver cirrhosis, and amenorrhea by ages 15–24. The type 2-associated cardiomyopathy is often irreversible despite initiation of phlebotomy or chelation therapy and may require an immediate transplant of the heart and potentially of the liver as well (von Herbay 1996, Jensen 1993).
Arthropathy. Joint changes in genetic haemochromatosis may occur in two different ways (Schuhmacher 1964, Dymock 1970, Niederau 1985, Niederau 1996). The most prevalent changes are seen in the metacarpophalangeal joints II and III, in the form of cystic and sclerotic changes, cartilage damage and a narrowing of the intraarticular space. Sometimes other joints of the hands and the feet are affected. Large joints, i.e., of the knees and hips, may be affected in the form of chondrocalcinosis. The pathogenesis of joint changes in haemochromatosis remains unclear. Arthropathy is one of the few complications not associated with the degree of iron overload. It has been speculated that iron may inhibit pyrophosphatase and may thereby lead to a crystallisation of calcium pyrophosphates. Alternatively, iron may have direct toxic effects on the joints. Arthropathy may be an early sign of haemochromatosis and may help to make the diagnosis at a precirrhotic stage (Niederau 1996). Haemochromatosis should therefore been considered in all patients with an arthropathy of unknown aetiology.
Endocrine abnormalities. In contrast to the early onset of arthropathic changes, endocrine abnormalities are a late consequence of iron overload. Sexual impotence and loss of libido may occur in up to 40% of male patients (Niederau 1985). The endocrine abnormalities in haemochromatosis are mainly, if not exclusively, due to pituitary failure. This is in contrast to alcoholic cirrhosis where testicular failure is predominant (Kley 1985a, Kley 1985b). In contrast to alcoholic cirrhosis, where oestrogen levels are usually increased, oestrogen levels were found decreased in haemochromatosis (Kley 1985a). Most endocrine changes are late and irreversible complications of genetic haemochromatosis and do not respond well to phlebotomy treatment (Niederau 1996). Iron overload only infrequently affects other endocrine organs such as the thyroid and adrenal glands. Severe hypogonadism with amenorrhea in young women and impotence in young men is today thought to be due to type 2 haemochromatosis.
Skin. Increased skin pigmentation is mainly seen in areas exposed to sunlight. A large part of the darkening of pigmentation is thought to be due to an increase in melanin and not due to iron excess itself. The increase in skin pigmentation is reversible on iron removal (i.e., phlebotomy).
Other potential complications. Iron overload has been speculated to aggravate atherosclerosis; however, the evidence for that is rather weak (for review see Niederau 2000). There have also been reports that extrahepatic malignancies may be increased in HFE haemochromatosis (Amman 1980, Fracanzani 2001) while other studies have not found extrahepatic associations (Bain 1984, Niederau 1996, Elmberg 2003). It is not clear whether HFE gene mutations are involved in the pathogenesis of porphyria cutanea tarda since the prevalence of both risk factors vary greatly in different parts of the world; associations between HFE gene mutations and porphyria have often been described in southern Europe but not in northern Europe (Toll 2006).
Polymorphisms beyond C282Y homozygocity. Recent studies have suggested that the C282Y and H63D polymorphisms in the HFE gene are associated with a selection advantage. This selection may also explain the high frequency of up to 40% of these polymorphisms seen in Celtic populations (Adams 2005). These polymorphisms are almost exclusively found in people with Celtic decent. A French study recently showed that these polymorphisms are seen in 27% of the French general population (Hermine 2015). Interestingly, 80% of French winners of WM, EM and Olympic sport events had one of these polymorphisms (Hermine 2015).
Along this line, a recent Swiss study showed that C282Y homozygotes are several centimeters taller than the reference population (Cippa 2013), although these homozygotes are usually considered not to be healthy. Indeed the greater height and physical fitness of the Celts have already been mentioned by Julius Caesar in his work „De Bello Gallico“ (Caesar 50 a.c.).
Thus, subjects with heterozygous HFE polymorphisms are usually “very healthy” people without a major risk for iron overload and associated organ damage. Only in the presence of other hepatotoxic factors such as hepatitis C or fatty liver disease HFE heterozygotes may have an increased risk to develop liver fibrosis (Erhardt 2003).
Therapy
Phlebotomy treatment. Phlebotomy treatment is the standard of care for removing iron in genetic haemochromatosis. One phlebotomy session removes approximately 250 mg iron from the body. Since patients with the classical clinical phenotype may have an excess of 10–30 g iron, it may take 12–24 months to remove the iron overload when phlebotomies of 500 mL blood are done weekly (Table 4). Phlebotomy treatment is generally well tolerated and hemoglobin usually does not drop below 12 g/dL. Several studies have shown that liver iron is completely removed at such low ferritin values; thus the effect of therapy can be checked by ferritin measurements and a control liver biopsy is not necessary. After complete removal of excess iron the intervals of phlebotomies may be increased to once every 2–3 months; serum ferritin should be kept in the lower normal range, between 50–100 ng/mL. Phlebotomy should not be interrupted for longer intervals; there is a risk of reaccumulation of iron due to the genetic autosomal recessive metabolic malfunction.
Erythrocytapheresis. Three prospective, randomised studies have compared the advantages and disadvantages of erythrocytapheresis compared to phlebotomy in patients with hereditary HFE haemochromatosis (Rombout-Sestrienkova 2012, Sundic 2013, Rombout-Sestrienkova 2016). Erythrocytapheresis can theoretically remove up to three times more red blood cells per single procedure when compared with regular phlebotomy and thus may have a clinical and economic benefit.
In one of these studies serum ferritin levels initially declined more rapidly in the apheresis group; however, time to normalisation of the ferritin level was equal in both groups (Sundic 2013). The cumulative costs for materials and technician times until achievement of the desired ferritin levels were three-fold higher in the apheresis group (Sundic 2013).
In the other study, after adjustments for initial serum ferritin and body weight, the number of therapeutic procedures was lower for erythrocytapheresis when compared with regular phlebotomy (0.43; 95% CI, 0.35–0.52; p <0.001) (Rombout-Sestrienkova 2012). Cost analysis however showed no significant difference in treatment costs between the two procedures (Rombout-Sestrienkova 2012).
The third study evaluated the effectiveness of erythrocytapheresis over phlebotomy for maintenance therapy in patients with HFE haemochromatosis (Rombout-Sestrienkova 2016). The two treatment-arms, randomised, crossover clinical trial involved 46 patients who were treated for one year with either erythrocytapheresis or phlebotomy to keep the ferritin level < 50 ng/mL. After one year, patients were switched to the other treatment modality. The mean number of treatment procedures per treatment year was significantly higher using phlebotomy versus erythrocytapheresis (3.3 vs. 1.9; p<0, 01). There was no significant difference between arms in overall health assessed by SF-36 and EQ-5D, respectively. The mean costs of one treatment year however were 235 € for phlebotomy versus 511 € for erythrocytapheresis.
In summary, regular phlebotomy remains the gold standard for removing excess iron in hereditary haemochromatosis type 1. It has few side-effects and is more cost-effective than erythrocytapheresis.
Monitoring of phlebotomy treatment. Phlebotomy treatment is usually monitored by repetitive measurements of serum ferritin. According to ESAL and AASLD guidelines, phlebotomies should be done at frequent intervals until serum ferritin is reduced to low normal values of about 50–100 ng/mL (Bacon 2011, EASL 2010). Thereafter, the interval of phlebotomies can be prolonged to assure that serum ferritin remains at 50–100 ng/mL. It is known that the liver and other organs do not contain excess iron when ferritin is in that range. On the other hand, it is also know that transferrin saturation may still be increased up to 70% at such ferritin levels in C282Y homozygotes. Recent studies have shown that serum concentrations of Non-Tranferrin-Bound Iron (NTBI) and Labile Plasma-Iron (LPI) may increase sharply beyond a tranferrin saturation of 70–80% (Cabantchik 2014). Such increases in NTBI and LPI may be associated with oxidative stress and risks for cell damage (Hershko 1978, Le Lan 2005, Pootrakul 2004, Hod 2011, Brissot 2012, Cabantchik 2014). Therefore, there is a current debate whether transferrin saturation should be used for monitoring long-term phlebotomy and transferrin saturation should aim to be kept below 50% (Cabantchik 2014, de Swart 2015). This would mean that a considerable number of patients would be at the risk to become iron deficient – which should be avoided according to EASL and AASLD guidelines (Bacon 2011, EASL 2010). It is also known that the usual ferritin monitoring assures a normal life expectancy in patients diagnosed without liver cirrhosis (Niederau 1985, Niederau 1996). Thus, as yet the monitoring of phlebotony treatment should be based on serum ferritin which should kept at 50–100 ng/mL (Bacon 2011, EASL 2010).
Iron removal by chelators. Deferoxamine therapy for genetic haemochromatosis is not recommended because phlebotomy is more effective with less side effects and lower cost. A phase 2/3 study proved the safety and effectiveness of the new oral iron chelator deferasirox in genetic HFE haemochromatosis (Phatak 2010). However, deferasirox is only currently approved for secondary haemochromatosis.
Diet. A diet low in iron is not recommended for patients with genetic haemochromatosis. One phlebotomy of 500 mL blood removes approximately 250 mg iron. A difficult-to-follow iron-restricted diet for a complete year would have the effect of a single phlebotomy. It is therefore recommended that patients simply do not eat excessive amounts of food with very high iron content (such as liver) and that they do not eat food to which iron has been added (Table 4). The recent EASL guidelines recommend to avoid supplemental vitamin C and to limit red meat and alcohol intake; cirrhotic patients should abstain from alcohol consumption (EASL 2022).
Use of proton pump inhibitors. A double-blind randomised placebo-controlled trial has shown that the use of proton pump inhibtors (PPI) significantly lowered the need for phlebotomies in the maintenance phase in patients with HFE haemochromatosis (Vanclooster 2017). This effect is probably mediated by diminishing the intestinal absorption of non-heme iron due to an increase in gastric pH. As stated by the recent EASL guideline (EASL 2022) PPI may have side effects and have not been studied in the induction phase of iron removal. Thus, PPI should be considered only as a supportive treatment in specific cases and not as a first- or second-line therapeutic tool (EASL 2022),
Liver transplantation. Advanced liver cirrhosis and carcinoma may be indications for a liver transplant in haemochromatosis (Kowdley 1995, Brandhagen 2000). The prognosis of patients who have a liver transplant for haemochromatosis is markedly worse than that for patients with other liver diseases; a considerable number of patients with haemochromatosis die after transplant from infectious complications or heart failure (Brandhagen 2000). Liver transplantation does not heal the original genetic defect.
Hepcidin administration. In HFE haemochromatosis hepcidin deficiency leads to increased intestinal absorption of dietary iron and subsequent to iron overload (Nemeth 2004). Thus, administration of hepcidin might be beneficial in this situation. However, hepcidin preparations turned out to have difficult pharmacokinetic characteristics which have recently been overcome by synthesis of the hepcidin mimetic rusfertide. Rusfertide has recently demonstrated control of iron in an animal model of hereditary haemochromatosis (Taranath 2022). In human HFE haemochromatosis rusfertide reduced transferrin saturation and serum iron with corresponding significant reduction in the number of phlebotomies in patients who had been on a stable phlebotomy regimen of 0.25 to 1 phlebotomy per month for at least 6 months (Kowdley 2021). Under the administration of rusfertide the liver iron concentration maintained at pre-study levels with minimal use of phlebotomies. Rusfertide was well tolerated by the patients (Kowdley 2021). These data suggest that rusfertide might be used as an alternative treatment for selected patients with HFE haemochromatosis in the future.
Prognosis
Untreated haemochromatosis often has a bad prognosis in the presence of liver cirrhosis and diabetes mellitus. The prognosis is markedly worse in patients with cirrhosis than in those without cirrhosis at diagnosis (Figure 3); the same is true for diabetes mellitus. It is generally accepted that phlebotomy therapy improves the prognosis. Patients diagnosed and treated in the early non-cirrhotic stage have a normal life expectancy (Figure 3) (Niederau 1985, Niederau 1996). Thus, early diagnosis markedly improves the prognosis (Figure 4). Iron removal by phlebotomy also improves the outcome in patients with liver cirrhosis. The prognosis of liver cirrhosis due to haemochromatosis is markedly better than those with other types of cirrhosis (Powell 1971). Hepatomegaly and elevation of aminotransferases often regress after iron removal (Niederau 1985, Niederau 1996) (Figure 5). Insulin-dependent diabetes mellitus and hypogonadism are irreversible complications despite complete iron removal (Niederau 1996) (Figure 5). Earlier changes in glucose and insulin metabolism, however, may be ameliorated after iron removal. For unknown reasons arthropathy does not respond well to phlebotomy treatment although it may be an early sign of iron overload (Figure 5). The AASLD consensus guidelines recommend to start phlebotomy treatment at ferritin values >300 ng/mL in men and >200 ng/mL in women. The risk for liver fibrosis and cirrhosis is increased only at ferritin levels >1000 ng/mL. Further studies need to determine whether asymptomatic C282Y homozygotes with ferritin values between 300 and 1000 ng/mL need to be treated or whether one might wait and monitor ferritin at that stage.
Juvenile hereditary haemochromatosis
Two genes have been associated with juvenile haemochromatosis: 90% of cases are associated with mutations in hemojuvelin (HJV) (locus name HFE2A, which encodes HJV), while 10% of cases are associated with HAMP (locus name HFE2B, which encodes hepcidin). Despite the nomenclature of HFE2A and HFE2B, juvenile haemochromatosis is not associated with HFE mutations. In order to avoid confusion most physicians use the terms type 2A (hemojuvelin mutations) and type 2B (HAMP mutations). Mutations in hemojuvelin are associated with low levels of hepcidin in urine suggesting that hemojuvelin regulates hepcidin. Hepcidin is the key regulator of intestinal iron absorption and iron release from macrophages. Hepcidin facilitates ferroportin internalisation and degradation. Hepcidin mutations may thereby lead to an increase in ferroportin and thus iron uptake from the intestine. Juvenile haemochromatosis is very rare. A clustering of HJV mutations can be seen in Italy and Greece although few families account for this phenomenon. Mutations in HJV represent the majority of worldwide cases of juvenile haemochromatosis.
Only a small number of patients have been identified with HAMP-related juvenile haemochromatosis. Juvenile haemochromatosis is characterised by an onset of severe iron overload in the first to third decades of life. Clinical features include hypogonadism, cardiomyopathy, and liver cirrhosis (Diamond 1989, Vaiopoulos 2003). The main cause of death is cardiomyopathy (De Gobbi 2002, Filali 2004). In contrast to HFE type 1 haemochromatosis, both sexes are equally affected. Mortality can be reduced in juvenile haemochromatosis when it is diagnosed early and treated properly. Phlebotomy is the standard therapy in juvenile haemochromatosis as well and is treated similarly to HFE haemochromatosis (Tavill 2001). In patients with juvenile haemochromatosis and anaemia or severe cardiac failure, administration of chelators such as deferoxamine have been tried to reduce mortality; some case reports suggest that this might improve left ventricular ejection fraction (Kelly 1998).
Transferrin receptor 2 (TFR2)-related type 3 haemochromatosis
TFR2-related haemochromatosis is defined as type 3 and is also known as HFE3; however, the term HFE3 should not be used because the HFE gene is not affected in type 3 haemochromatosis. TFR2-related haemochromatosis is inherited in an autosomal recessive manner. TFR2 is a type II 801-amino acid transmembrane glycoprotein expressed in hepatocytes and at lower levels in Kupffer cells (Zhang 2004). A finely regulated interaction between TFR2, TFR1 and HFE is now thought to affect the hepcidin pathway, and, consequently, iron homeostasis (Fleming 2005). Patients with homozygous TFR2 mutations have increased intestinal iron absorption that leads to iron overload. Hepcidin concentrations in urine are low in TFR2 haemochromatosis (Nemeth 2005). TFR2-related haemochromatosis is very rare with only about 20 patients reported worldwide (Mattman 2002). Age of onset in TFR2-related type 3 haemochromatosis is earlier than in HFE-associated type 1 (Piperno 2004, Girelli 2002, Hattori 2003). Progression is, however, slower than in juvenile type 2 (De Gobbi 2002, Roetto 2001, Girelli 2002). The phenotype is similar to type 1. Many patients present with fatigue, arthralgia, abdominal pain, decreased libido, or with biochemical signs of iron overload (Roetto 2001, Girelli 2002, Hattori 2003). Complications of type 3 haemochromatosis include cirrhosis, hypogonadism, and arthropathy. Cardiomyopathy and diabetes mellitus appear to be rather rare. Hepatocellular carcinoma has not been observed in the small number of cases diagnosed. Most individuals with type 3 haemochromatosis have an Italian or Japanese genetic background. Some of the Japanese males have had liver cirrhosis at diagnosis (Hattori 2003). Similar to type 1 haemochromatosis, the penetration of type 3 haemochromatosis is also considerably less than 100% (Roetto 2001). Standard therapy is iron removal by weekly phlebotomy similar to the management of type 1 disease. Individuals with increased ferritin should be treated similar to those with HFE haemochromatosis.
Type 4 haemochromatosis – Ferroportin Disease
Ferroportin-associated iron overload (also called Ferroportin Disease) was first recognised by Pietrangelo (1999) who described an Italian family with an autosomal dominant non-HFE haemochromatosis. Many family members had iron overload resulting in liver fibrosis, diabetes, impotence, and cardiac arrhythmias. In addition to autosomal dominant inheritance, features distinguishing this from HFE haemochromatosis included early iron accumulation in reticuloendothelial cells and a marked increase in ferritin earlier than what is seen in transferrin saturation (Pietrangelo 1999, Rivard 2003, Montosi 2001, Wallace 2004, Fleming 2001). Several patients showed a reduced tolerance to phlebotomy and became anemic despite elevated ferritin (Pietrangelo 1999, Jouanolle 2003).
In 2001, this form of non-HFE haemochromatosis was linked to mutations of ferroportin (Montosi 2001) that had just been identified as the basolateral iron transporter (Abboud 2000, Donovan 2000). Since that time, numerous mutations in the gene have been implicated in patients from diverse ethnic origins with previously unexplained haemochromatosis. Iron overload disease due to ferroportin mutations has been defined as type 4 haemochromatosis or Ferroportin Disease (for review see Pietrangelo 2004 and 2017). The iron export is tightly regulated because both iron deficiency and iron excess are harmful. The main regulator of this mechanism is the peptide hepcidin which binds to ferroportin, induces its internalisation and degradation, thereby reducing iron efflux (Nemeth 2004). Increase in iron absorption may be caused either by hepcidin deficiency or its ineffective interaction with ferroportin. All recent studies have shown that hepcidin deficiency appears to be the common characteristic of most types of genetic haemochromatosis (mutations in HFE, transferrin receptor 2, hemojuvelin, or hepcidin itself). The remaining cases of genetic iron overload are due to heterozygous mutations in the hepcidin target, ferroportin. Because of the mild clinical penetrance of the genetic defect there were doubts about the rationale for iron removal therapy. However, a more recent study shows that there may be clinically relevant iron overload with organ damage and liver cancer in patients carrying the A77D mutation of ferroportin (Corradini 2007). Treatment schemes are similar to those described for other types of genetic haemochromatosis.
Loss-of-function of one ferroportin 1 allele results in haemochromatosis type 4a with a normal intestinal iron export but a diminished iron export from tissue macrophages leading to progressive iron accumulation in liver, spleen and bone marrow macrophages and in an inappropriately low iron delivery to circulating transferrin (Gozzelino 2016). Thus in the presence of high serum ferritin, transferrin saturation is relatively low in this type of genetic iron overload. The diminished iron delivery may also result in reduced erythropoiesis and anaemia. Liver iron deposition is primarily seen in macrophages (Wallace 2013).
The rare gain-of-function mutations of ferroportin can lead to haemochromatosis type 4b by reducing the inhibitory activity of hepcidin leading to an increase in intestinal iron absorption and to a release of iron from macrophages. Transferrin saturation is high (Callebau 2014). Liver iron deposition is primarily seen in hepatocytes (Kasvosv 2013).
Secondary haemochromatosis
Pathophysiology
Most forms of secondary haemochromatosis are due to hemolytic anaemia associated with polytransfusions such as thalassaemia, sickle cell disease, and myelodysplastic syndromes (MDS). Most of these patients need blood transfusions on a regular basis for survival. However, in the long run, multiple blood transfusions often lead to iron overload if patients are not treated with iron chelators. In general, iron overload due to blood transfusions is similar to genetic haemochromatosis; however, secondary iron overload develops much faster than the genetic forms (McLaren 1983), sometimes as soon as after 10–12 blood transfusions (Porter 2001). Subsequently secondary iron overload can result in more rapid organ damage when compared with genetic haemochromatosis. Secondary iron overload can obviously not be treated by phlebotomy because a marked anaemia is the clinical marker of the disease. Secondary iron overload often limits the prognosis of patients with thalassaemia; life expectancy deteriorates with increasing iron concentrations in the liver (Telfer 2000). Therapy with iron chelator may reduce the transfusional iron burden if the frequency of transfusion is not too high. The development of HFE versus secondary haemochromatosis not only differs in terms of the speed of iron accumulation but also in the type of organ damage; in secondary haemochromatosis cardiomyopathy is often the complication that limits the prognosis (Liu 1994). It is interesting that heart disease is also very frequent in juvenile genetic haemochromatosis where there is also rapid iron accumulation. In general, serum ferritin values closely reflect liver iron concentration and may be used as an indication for timing of therapy as well as to check the effects of iron chelation.
For many years, deferoxamine was the only iron chelator available in most countries but in some countries deferiprone is also approved for patients who do not tolerate deferoxamine (Hoffbrandt 2003). The clinical use of deferiprone is limited due to side effects such as agranulocytosis and neutropenia (Refaie 1995). Long-term data prove that deferoxamine can reduce iron overload and its organ complications (Olivieri 1994, Cohen 1981). Deferoxamine, however, needs to be given daily subcutaneously or by IV infusion for several hours. Thus, patients with thalassaemia often report that deferoxamine treatment is worse than thalassaemia itself (Goldbeck 2000). Therefore, adherence problems often limit the beneficial effects of this iron chelator (Cohen 1989).
Without iron chelation, children with thalassaemia often develop a severe cardiomyopathy prior to age 15 (Cohen 1987). After that age, liver cirrhosis is also a significant complication in secondary iron overload due to thalassaemia (Zurlo 1992). Iron chelation should start early to prevent complications of iron overload. By the ages of 3–5, liver iron concentration may reach values associated with a significant risk for liver fibrosis in severe thalassaemia (Angelucci 1995). Children younger than 5 should therefore be cautiously treated with chelators if they have received transfusions for more than a year (Olivieri 1997). Deferoxamine can reduce the incidence and ameliorate the course of iron-associated cardiomyopathy (Olivieri 1994, Brittenham 1994, Miskin 2003).
Deferasirox is an oral iron chelator with high selectivity for iron III (Nick 2003). Deferasirox binds iron in a 2:1 proportion with a high affinity and increases the biliary iron excretion (Nick 2003). This chelator is able to reduce iron overload in hepatocytes and cardiomyocytes (Nick 2003, Hershko 2001). Due to its half-life of 11–18 hours it needs to be taken only once daily (Nisbet-Brown 2003). Deferasirox exerted a similar iron chelation when compared with deferoxamine in patients with thalassaemia; the effect of 40 mg/kg deferoxamine was similar to that of 20 mg/kg deferasirox (Piga 2006). Both in adults and children 20–30 mg/kg/day deferasirox significantly reduced liver iron concentration and serum ferritin (Cappellini 2006). Magnetic resonance imaging showed that 10–30 mg/kg/day deferasirox may also reduce iron concentration in the heart within one year of maintenance therapy. Deferasirox may cause minor increases in serum creatinine as well as gastrointestinal discomfort and skin exanthema which are usually self-limiting. Considering the compliance problems with deferoxamine, deferasirox has a better cost-effectiveness ratio (Vichinsky 2005). Deferasirox is defined as standard therapy both in the guidelines of the National Comprehensive Cancer Network (NCCN) (USA) and in the international guidelines on MDS (Greenberg 2006, Gattermann 2005).
Use of blood from patients with HFE haemochromatosis (type 1) for blood donation
For some decades it has been debated whether blood phlebotomised from patients with HFE haemochromatosis may be used for blood transfusions (Nouel 1991, Barton 1999, Conry-Cantilena 2001, De Buck 2012, Leitmann 2013). In many countries blood from haemochromatosis patients is still not used for blood transfusion because of several arguments and precautions:
For a long time such blood has not been accepted by many blood banks because there was a hypothesis that such blood may be associated with increased risk for the recipient. Indeed, excess iron may increase the risk for bacterial and viral infections (Walker 2000, Khan 2007, Drakesmith 2008). In particular there were some hints that siderophilic bacteria including Vibrio sp., Salmonella sp. and Yersinia sp. grow particularly well in iron-overloaded blood (Nouel 1991, Cauchie 1987, Boelaert 1987, Piroth 1997). There have also been reports that Yersinia enterocolitica is responsible for posttransfusion sepsis and death (Leclercq 2005). In vitro there is a significantly decreased antibacterial activity against S. typhimurium LT2 and a better survival of Vibrio vulnificus in blood from iron-overloaded HFE patients when compared with healthy subjects (Jolivet-Gougeon 2007, Jolivet-Gougeon 2008, Bullen 1991).
In contrast, such risks were not present in blood from iron-depleted patients with HFE haemochromatosis (Jolivet-Gougeon 2008, Bullen 1991). A further study showed that the presence of anti-Yersinia antibodies was similar in the blood of uncomplicated HFE haemochromatosis patients when compared to blood from control donors (Jolivet-Gougeon 2007). Based on screening tests for antibodies to hepatitis B core antigen, syphilis, human immunodeficiency virus, hepatitis C virus, hepatitis B surface antigen, and human T-lymphotropic virus, no statistically significant difference could be found for HFE donors versus regular donors (Leitman 2003, Sanchez 2001).
It has in addition been argued that the blood donation by haemochromatosis patients is not voluntary because they benefit from the donation (Conry-Cantilena 2001, De Gonzalez 2007, Pennings 2005). Also phlebotomies from haemochromatosis patients does not require a financial compensation and may thus provide a financial advantage for the physician (Leitman 2013). The latter argument needs to be discussed considering that management of haemochromatosis patients as well as the use of their blood vary between industrialised countries (Butzeck 2011, Leitman 2013). In any case, it has been proposed that all phlebotomies should be free to haemochromatosis patients in order to eliminate any financial incentives and the non-voluntary character of the donation (Leitman 2013).
In general, blood banks need to observe rigorously that their criteria for haemochromatosis patients are also applicable to other donors. In a cohort of 130 subjects with HFE polymorphisms referred to a blood centre for management, 76% met all eligibility criteria for allogeneic blood donation and 55% had previously been blood donors before being made aware of their HFE diagnosis (Leitmann 2003). In the latter study, HFE donors were documented to more regularly observing their donation appointments than non-HFE donors, and they were less likely to have low screening hemoglobin of < 12.5 g/dL (Leitman 2003).
Since 2001, many European and U.S. transfusion services have changed their policy for the management of blood drawn from haemochromatosis patients (Courtois 2001, Radojska 2011, Buring 2002, Guidelines for the Blood Transfusion Services in the United Kingdom 2005, Ministrial Order of the Government of France 2009, FDA guidance for variances for blood collection from individuals with hereditary haemochromatosis 2001). For the USA, the FDA (Food and Drug Administration) issued a guidance in 2001 to allow blood banks to submit variances to federal code to accept blood from HFE patient for blood transfusion (Center for Biologics Evaluation and Research 2013). This guidance contains several criteria (Leitman 2013):
- The donor meets all other general allogeneic donor criteria.
- Phlebotomy is provided free of charge to all HFE patients in that blood centre.
- Incentives for HFE donors are considered untruthful in responding to standardised health history screening questions.
- A medical prescription for phlebotomy therapy including frequency and hemoglobin threshold is provided by the donor’s physician.
- A short physical examination is performed at each visit if the patient donates more often than every 8 weeks.
In the 12 years following the publication of this guidance, 163 blood banks in 43 US states have submitted variances and implemented polices for collection of blood from HFE donors (Leitman 2013). HFE donors have been shown to have a considerable satisfaction from knowing that their blood is being used to save lives rather than being discarded (Center for Biologics Evaluation and Research 2013).
It is estimated that routine referral of HFE subjects to blood centres for phlebotomy care could supplement the U.S. blood supply by an additional 1.3 million RBC units per year, possibly help to avoid periodic blood shortages, avoid wastage of safe units, and decrease the costs of care (Leitman 2013).
People with C282Y/H63D and H63D/H63D genotypes and slightly elevated ferritin levels are often referred to the blood centre for phlebotomy treatment (Leitman 2013). These subjects in general do not have organ damage due to iron overload and do need an aggressive phlebotomy therapy like the C282Y homozygotes. In blood centres with active recruitment of HFE patients, blood donations from HFE patients may contribute to 10 – 40% of available blood (Leitman 2013).
Nevertheless, there is still no consensus about the acceptance of haemochromatosis patients as blood donors (Leitman 2013, de Buck 2012). Most recent studies however share the following policy when dealing with a potential acceptance of haemochromatosis patients as blood donors (De Buck 2012, Sackett 1996):
In general, all criteria applicable to any other donor need to be rigorously observed also for HFE patients. Blood from HFE patients should only be used for transfusion when patients have already been iron-depleted and do not have major organ complications. There are no incentives or financial advantages for the HFE patients and their physicians for the use of phlebotomised blood for donation.
Key messages and future directions
- Measurement of transferrin saturation and serum ferritin is the first and most important step in the diagnosis of genetic haemochromatosis. High serum ferritin with normal transferrin saturation is usually not associated with genetic haemochromatosis (except for the rare ferroportin type 4b disease).
- Individuals with signs of iron overload, females with transferrin saturation >45% and serum ferritin > 200µg/L and males with transferrin saturation >50% and ferritin >300µg/L, or otherwise unexplained high transferrin saturation should be tested for the C282Y variation in the HFE gene. The search for other HFE gene variations is less important.
- The homozygous C282Y mutation accounts for <90% of phenotypes in Caucasians and leads to an increase in intestinal iron absorption with a risk of iron overload and organ damage including liver cirrhosis and diabetes mellitus.
- Early diagnosis in a non-cirrhotic stage and subsequent iron removal by phlebotomies are associated with a normal life expectancy. All future efforts should aim at such early diagnosis.
- All patients with haemochromatosis should be assessed for liver fibrosis. In patients with ferritin <1000 µg/L and no signs of liver disease, the risk of fibrosis is very low. Elastography can rule out advanced fibrosis. Liver biopsy is only necessary when fibrosis stage remains unclear; liver iron concentration should rather be assessed by MRI and not by biopsy.
- Patients with cirrhosis or severe fibrosis have a risk for development of HCC and should be offered adequate HCC surveillance.
- Young individuals with evidence of haemochromatosis (amenorrhea, hypogonadism, cardiomyopathy) should be tested for rare haemochromatosis gene variants. Patients with evidence of significant, unexplained iron overload should be referred to an iron disorder specialist to look for further rare genetic defects associated with haemochromatosis (e.g. HFE, HAMP, HJV, TFR2, TF, CP, BMP6, SCL40A1).
- Secondary haemochromatosis is usually caused by multiple blood transfusions in haemolytic anaemias such as thalassaemia, sickle cell anaemia, and myelodysplasia. Here, iron may accumulate faster than in genetic haemochromatosis with risks for cardiomyopathy and liver cirrhosis. Therapy consists of iron chelators because phlebotomies cannot be done due to underlying anaemia.
References
Abboud S, Haile DJ. A novel mammalian iron-regulated protein involved in intracellular iron metabolism. J Biol Chem 2000;275:19906-12.
Adams PC, Speechley M, Kertesz AE. Long-term survival analysis in hereditary haemochromatosis. Gastroenterology 1991;101:368-72.
Adams PC, Reboussin DM, Barton JC, et al. Haemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med 2005; 352:1769-78.
Andriopoulos B Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet 2009;41:482-7.
Angelucci E, Baronciani D, Lucarelli G, et al. Needle liver biopsy in thalassaemia: analyses of diagnostic accuracy and safety in 1184 consecutive biopsies. Br J Haematol 1995;89:757-61.
Bacon BR, Adams PC, Kowdley KV, Powell LW, Tavill AS. Diagnosis and management of haemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology 2011;54:328-43.
Bain C, Bradbear R, Siskind V, et al. Cohort study of the risk of malignancy in haemochromatosis and other nonalcoholic liver diseases. Hepatology 1984;4:A1020.
Barton JC, Grindon AJ, Barton NH, Bertoli LF. Haemochromatosis probands as blood donors. Transfusion 1999;39:578-85.
Biasiotto G, Belloli S, Ruggeri G, et al. Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin Chem 2003;49:1981-8.
Bierens deHaan B, Scherrer JR, Stauffacher W, et al. Iron excess, early glucose intolerance, and impaired insulin secretion in idiopathic haemochromatosis. Eur J Clin Invest 1973;3:179-87.
Blei AT, Robbins DC, Drobny E, et al. Insulin resistance and insulin receptors in hepatic cirrhosis. Gastroenterology 1982;83:1313-8.
Boelaert JR, van Landuyt HW, Valcke YJ, et al. The role of iron overload in Yersinia enterocolitica and Yersinia pseudotuberculosis bacteraemia in hemodialysis patients. J Infect Dis 1987;156:384-7.
Brandhagen DJ, Alvarez W, Therneau TM, et al. Iron overload in cirrhosis-HFE genotypes and outcome after liver transplantation. Hepatology 2000;31:456-60.
Brissot P, Ropert M, Le Lan C, Loreal O. Non-transferrin bound iron: a key role in iron overload and iron toxicity. Biochim Biophys Acta 2012;1820:403-10.
Brittenham GM, Griffith PM, Nienhuis AW, et al. Efficacy of deferoxamine in preventing complications of iron overload in patients with thalassaemia major. N Engl J Med 1994;331:567-73.
de Buck E, Pauwels NS, Dieltjens T, et al. Is blood of uncomplicated haemochromatosis patients safe and effective for blood transfusion? A systematic review J Hepatol 2012; 57: 1126-34.
Buja L, Roberts W. C Iron in the heart. Am J Med 1971;51:209-21.
Buring ML. Haemochromatosis: red cross blood service policy. Med J Aust 2002;176:564.
Bullen JJ, Spalding PB, Ward CG, Gutteridge JMC. Haemochromatosis, iron, and septicaemia caused by Vibrio vulnificus. Arch Intern Med 1991;151:1606-9.
Butzeck B, Rialland J, Brissot P, Porto G, Courtois F. Management, costs of phlebotomies and use of removed blood in haemochromatosis varies throughout Europe. Am J Hematol 2011;86:E81.
Cabantchik ZI. Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Frontiers Pharmacol 2014;5:1-11.
Callebaut I, Joubrel R, Pissard S, et al. Comprehensive functional annotationof 18 missense mutations found in suspected haemochromatosis type 4 patients. Human Mol Genet 2014; 23:4479-90.
Caesar J. Commentarii de Bello Gallico, Book II, Chapter 30. 50 a.c. www.thelatinlibrary.com/caesar/gall1.shtml
Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassaemia. Blood 2006;107:3455-62.
Cauchie P, Vincken W, Peeters O, Charels K. Haemochromatosis and Yersinia enterocolitica septicaemia. Dig Dis Sci 1987;32:1438.
Center for Biologics Evaluation and Research. Guidance for industry: variances for blood collection from individuals with hereditary haemochromatosis. Available from: http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/blood/ucm076719.htm. Accessed May 13, 2013.
Cippà PE, Krayenbuehl P. Increased height in HFE haemochromatosis. N Engl J Med 2013;369:785-6.
Conry-Cantilena C. Phlebotomy, blood donation, and hereditary haemochromatosis.Transfus Med Rev 2001;15:136-43.
Courtois F, Danic B. Genetic haemochromatosis and blood donation. Ann Med Interne 2001;152:452-54.
Cohen A, Martin M, Schwartz E. Response to long-term deferoxamine therapy in thalassaemia. J Pediatr 1981;99:689-94.
Cohen AR, Mizanin J, Schwartz E. Rapid removal of excessive iron with daily, high-dose intravenous chelation therapy. J Pediatr 1989;115:151-5.
Cohen A. Management of iron overload in the paediatric patient. Hematol Oncol Clin North Am 1987;1:521-44.
Corradini E, Ferrara F, Pollicino T, et al. Disease progression and liver cancer in the ferroportin disease. Gut 2007;56:1030-2.
Creutzfeldt W, Frerichs H, Sickinger K. Liver diseases and diabetes mellitus. Prog Liver Dis 1970;3:371-407.
Crosby WH. Hereditary haemochromatosis. In: Ingelfinger FJ (ed) Controversy in internal medicine. Saunders, Philadelphia, 1966:261-70.
Daher R, Kannengiesser C, Houamel D, et al. Heterozygous Mutations in BMP6 pro-peptide lead to inappropriate hepcidin synthesis and moderate iron overload in humans. Gastroenterology 2016;150:672-83.
Datz C, Müller E, Aigner E. Iron overload and non-alcoholic fatty liver disease. Minerva Endocrinol 2017;42:173-83.
De Domenico I, Diane M, Ward DM, et al. Hepcidin regulation: ironing out the details. J Clin Invest 2007;117:1755-8.
De Gobbi M, Pasquero P, Brunello F, et al. Juvenile haemochromatosis associated with B-thalassaemia treated by phlebotomy and recombinant human erythropoietin. Haematologica 2000;85:865-7.
De Gonzalez GL. Hereditary haemochromatosis and blood donation. ISBT Sci Ser 2007;2:12-8.
Deugnier Y, Bardou-Jacquet É, Lainé F. Dysmetabolic iron overload syndrome (DIOS). Presse Med 2017;46: e306-11.
Diamond T, Stiel D, Posen S. Osteoporosis in haemochromatosis: iron excess, gonadal deficiency, or other factors? Ann Intern Med 1989;110:430-6.
Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature 2000;403:778-81.
Doyle A, Rusli F, Bhathal P. Aceruloplasminaemia: a rare but important cause of iron overload. BMJ Case Repn2015: bcr2014207541.
Drakesmith H, Prentice A. Viral infection and iron metabolism. Nat Rev Microbiol 2008;6:541–52.
Dymock W, Hamilton EBD, Laws JW, et al. Arthropathy of haemochromatosis: clinical and radiological analysis of 73 patients with iron overload. Ann Rheum Dis 1970;29:469-76.
Dymock W, Cassar J, Pyke DA, et al. Observations on the pathogenesis, complications, and treatment of diabetes in 115 cases of haemochromatosis. Am J Med 1972;52:203-10.
EASL. EASL clinical practice guidelines for HFE haemochromatosis. J Hepatol 2010; 53:3-22.
European Association for the Study of the Liver. EASL Clinical practice guidelines on haemochromatosis. J Hepatol 2022;77:479-502
Edwards CQ, Griffen LM, Goldgar D, et al. Prevalence of haemochromatosis among 11, 065 presumably healthy blood donors. N Engl J Med 1988;318:1355-62.
Elmberg M, Hultcrantz R, Ekbom A, et al. Cancer risk in patients with hereditary haemochromatosis and in their first-degree relatives. Gastroenterology 2003;125:1733-41.
Erhardt A, Niederau C, Osman Y, et al. Demonstration of HFE polymorphism in German patients with hereditary haemochromatosis. Dtsch Med Wochenschr 1999;124:1448-52.
Erhardt A, Maschner-Olberg A, Mellenthin C, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol 2003;38:335-42.
Fargion S, Mandelli C, Piperno A, et al. Survival and prognostic factors in 212 Italian patients with genetic haemochromatosis. Hepatology 1992;15:655-9.
Feder JN, Gnirke A, Thomas W, et al. A novel MCH class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics 1996;13:399-407.
Feldman AG, Whitington PF. Neonatal haemochromatosis. J Clin Exp Hepatol 2013;3:313-20.
Fernandez M, Lokan J, Leung C, Grigg A. A critical evaluation of the role of iron overload in fatty liver disease. J Gastroenterol Hepatol 2022;37:1873-83.
Filali M, Le Jeunne C, Durand E, et al. Juvenile haemochromatosis HJV-related revealed by cardiogenic shock. Blood Cells Mol Dis 2004;33:120-4.
Finch SC, Finch CA. Idiopathic haemochromatosis, an iron storage disease. Medicine 1966;34:381-430.
Fleming RE, Sly WS. Ferroportin mutation in autosomal dominant haemochromatosis: loss of function, gain in understanding. J Clin Invest 2001;108:521-2.
Fleming RE, Sly WS. Mechanisms of iron accumulation in hereditary haemochromatosis. Ann Rev Physiol 2002;4:663-80.
Fletcher LM, Halliday JW. Haemochromatosis: Understanding the mechanism of disease and implications for diagnosis and patient management following the recent cloning of novel genes involved in iron metabolism. J Intern Med 2002;251:181-92.
Gallego CJ, Burt A, Sundaresan AS, et al. Penetrance of haemochromatosis in HFE genotypes resulting in p.Cys282Tyr and p.[Cys282Tyr];[His63Asp] in the eMERGE network. Am J Hum Genet 2015;97:512-20.
Gattermann N. Consensus statement on iron overload in myelodysplastic syndromes. Hematol Oncol Clin N Am 2005;19:S18-S25.
Girelli D, Bozzini C, Roetto A. Clinical and pathological finding in haemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology 2002;122:1295-1302.
Goldbeck L, Baving A, Kohne E. Psychosocial aspects of beta-thalassaemia: distress, coping and adherence. Klin Pädiatr 2000;212:254-9.
Gozzelino R, Arosio P. Iron homeostasis in health and disease. Intern J Mol Sci 2016;17:130.
Greenberg PL, Baer MR, Bennett JM, et al. Myelodysplastic syndromesclinical practice guidelines in oncology. J Natl Compr Canc Netw 2006;4:58-77.
Guidelines for the Blood Transfusion Services in the United Kingdom. seventh ed. London: TSO (The Stationary Office); 2005.
Hanot V, Schachmann M. Sur la cirrhose pigmentaire dans le diabe`te. sucré. Arch Physiol Norm Pathol 1886;7:50-72.
Hardy L, Hansen JL, Kushner JP, Knisely AS. Neonatal haemochromatosis. Genetic analysis of transferrin-receptor, H-apoferritin, and L-apoferritin loci and of the human leukocyte antigen class I region. Am J Pathol 1990;137:149-53.
Hattori A, Wakusawa S, Hayashi H, et al. AVAQ 594-597 deletion of the TfR2 gene in a Japanese family with haemochromatosis. Hepatol Res 2003;26:154-6.
Herbay von A, Niederau C, Pelichowska M, et al. Kardiomyopathie als Todesursache bei genetischer Hämochromatose. Z Gastroenterol 1996;34:178-82.
Hermine O, Dine G, Genty V, et al. Eighty percent of French sport winners in Olympic, World and Europeans competitions have mutations in the haemochromatosis HFE gene. Biochimie 2015;119:1-5.
Hershko C, Graham G, Bates GW, Rachmilewitz EA. Non-specific serum iron in thalassaemia: an abnormal serum iron fraction of potential toxicity. Br J Haematol 1978;40:255-63.
Hershko C, Konijn AM, Nick HP, et al. ICL670A: a new syntheticoral chelator: evaluation in hypertransfused rats with selective radio iron probes of hepatocellular and reticuloendothelial iron stores and in ironloadedrat heart cells in culture. Blood 2001 ;97:1115-22.
Hider RC, Silva AM, Podinovskaia M, Ma Y. Monitoring the efficiency of iron chelation therapy: the potential of nontransferrin-bound iron. Ann N Y Acad Sci 2010;1202:94-9.
Hod EA, Brittenham GM, Billote GB, et al. Transfusion of human volunteers with older, stored red blood cells produces extravascular hemolysis and circulating non-transferrin bound iron. Blood. 2011;118:6675-82.
Jensen PD, Bagger JP, Jensen FT, et al. Heart transplantation in a case of juvenile hereditary haemochromatosis followed up by MRI and endomyocardial biopsies. Eur J Haematol 1993;51:199-205.
Jolivet-Gougeon A, Loreal O, Ingels A, et al. Serum transferrin saturation increase is associated with decrease of antibacterial activity of serum in patients with HFE-related genetic haemochromatosis. Am J Gastroenterol 2008;103:2502–2508.
Jolivet-Gougeon A, Ingels A, Danic B, et al. No increased seroprevalence of anti-Yersinia antibodies in patients with type 1 (C282Y/C282Y) haemochromatosis. Scand J Gastroenterol 2007;42: 1388–9.
Jouanolle AM, Douabin-Gicquel V, Halimi C, et al. Novel mutation in ferroportin 1 gene is associated with autosomal dominant iron overload. J Hepatol 2003;39:286-9.
Kasvosve I. Effect of ferroportin polymorphism on iron homeostasis and infection. Clinica Chimica Acta 2013;416:20-25.
Kelly AL, Lunt PW, Rodrigues F, et al. Classification and genetic features of neonatal haemochromatosis: a study of 27 affected pedigrees and molecular analysis of genes implicated in iron metabolism. J Med Genet 2001;38:599-610.
Khan FA, Fisher MA, Khakoo RA. Association of haemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis 2007;11:482-7.
Kelly AL, Rhodes DA, Roland JM, et al. Hereditary juvenile haemochromatosis: a genetically heterogeneous life-threatening iron-storage disease. QJM 1998;91:607-18.
Kley HK, Stremmel W, Niederau C, et al. Androgen and oestrogen response to adrenal and gonadal stimulation in idiopathic haemochromatosis: evidence for decreased oestrogen formation Hepatology 1985a:251-6.
Kley HK, Niederau C, Stremmel W, et al. Conversion of androgens to oestrogens in idiopathic haemochromatosis: comparison with alcoholic cirrhosis. J Clin Endocrinol Metabol 1985b;61:1-6.
Kowdley K, Hassanein T, Kaur S, et al. Primary liver cancer and survival in patients undergoing liver transplantation for haemochromatosis. Liver Transpl Surg 1995;1:237-41.
Kowdley KV, Modi NB, Valone F, et al. Rusfertide (PTG-300), a hepcidin mimetic, maintains liver iron concentration in the absence of phlebotomies in patients with hereditary haemochromatosis. Blood 2021:13:S943.
Leclercq A, Martin L, Vergnes ML, et al. Fatal Yersinia enterocolitica biotype 4 serovar O:3 sepsis after red blood cell transfusion. Transfusion 2005;45:814–8.
Leitman SF. Haemochromatosis: the new blood donor. Transfusion Med 2013; 645-50.
Leitman S, Browning J, Yau Y, et al. Haemochromatosisc subjects as allogeneic blood donors: a prospective study. Transfusion 2003;43:1538-44.
Le Lan C, Loreal O, Cohen T, et al. Redox active plasma iron in C282Y/C282Y haemochromatosis. Blood 2005;105:4527-31.
Liu P, Olivieri N. Iron overload cardiomyopathies: new insights into an old disease. Cardiovasc Drugs Ther 1994;8:101-10.
Loreal O, Deugnier Y, Moirand R. Liver fibrosis in genetic haemochromatosis. Respective roles of iron and non-iron related factors in 127 homozygous patients. J Hepatol 1992;16:122-7.
MacDonald RA, Mallory GK. Haemochromatosis and hemosiderosis. Study in 211 autopsied cases. Arch Intern Med 1960;105:686-700.
Mattman A, Huntsman D, Lockitch G, et al. Transferrin receptor 2. (TfR2) and HFE mutational analysis in non-C282Y iron overload: identification of a novel TfR2 mutation. Blood 2002;100:1075-7.
McLaren GD, Muir WA, Kellermeyer RW. In overload disorders: natural history, pathogenesis, diagnosis, and therapy. Crit Rev Clin Lab Sci 1983;19:205-66.
Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload Nat Genet 2009;41:478-81.
Ministrial Order of the Government of France. Arrêté du 12 janvier 2009 fixants les critères de selection des donneurs de sang 2009.
Miskin H, Yaniv I, Berant M, et al. Reversal of cardiac complications in thalassaemia major by long-term intermittent daily intensive iron chelation. Eur J Haematol 2003;70:398-403.
Montosi G, Donovan A, Totaro A, et al. Autosomal-dominant haemochromatosis is associated with a mutation in the ferroportin (SLC11A3) gene. J Clin Invest 2001;108:619-3.
Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalisation. Science 2004;306:2090-3.
Nemeth E, Roetto A, Garozzo G, et al. Hepcidin is decreased in TFR2-Haemochromatosis. Blood 2005;105:1803-6.
Newman B. Haemochromatosis blood donor programmes: marginal for the red blood cell supply but potentially good for patient care. Transfusion 2004;44:1535-7.
Nick H, Acklin P, Lattmann R, et al. Development of tridentate iron chelators: from desferrithiocin to ICL670. Curr Med Chem 2003;10:1065-76.
Niederau C, Berger M, Stremmel W, et al. Hyperinsulinaemia in non-cirrhotic haemo-chromatosis: impaired hepatic insulin degradation? Diabetologia 1084; 26:441-4.
Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and noncirrhotic patients with primary haemochromatosis. New Engl J Med 1985;313:1256-62.
Niederau C, Fischer R, Pürschel A, et al. Long-term survival in patients with hereditary haemochromatosis. Gastroenterology 1996;110:1107-19.
Niederau C, Niederau CM, Littauer A, et al. Screening for iron overload and iron deficiency. Ann Int Med 1998;128:337-45.
Niederau C. Diabetes mellitus bei Hämochromatose. Z Gastroenterol 1999;37:22-32.
Niederau C. Hepatische Eisenüberladung. Gastro-News 2018;5:46-53.
Niederau C. Iron overload and atherosclerosis. Hepatology 2000;32:672-4.
Nisbet-Brown E, Olivieri F, Giardina PJ, et al. Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: a randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2003;361:1597-602.
Njajou OT, Vaessen N, Joosse M, et al. A mutation in SLC11A3 is associated with autosomal dominant haemochromatosis. Nat Genet 2001;28:213-4.
Nouel O, Voisin PM, Vaucel J, Dartois-Hoguin M, Le BM. Yersinia enterocolitica septicaemia associated with idiopathic haemochromatosis and deferoxamine therapy. A case. Presse Med 1991;20:1494-6.
Olivieri NF, Brittenham GM. Iron-chelating therapy and the treatment of thalassaemia. Blood 1997;89:739-61.
Olivieri NF, Nathan DG, Macmillan JH, et al. Survival in medically treated patients with homozygous beta-thalassaemia. N Engl J Med 1994;331:574-8.
Pennings G. Demanding pure motives for donation: the moral acceptability of blood donations by haemochromatosis patients. J Med Ethics 2005;31:69-72.
Phatak PD, Sham RL, Raubertas RF, et al. Prevalence of hereditary haemochromatosis in 16, 031 primary care patients. Ann Intern Med 1998;129:954-61.
Phatak P, Brissot P, Wurster M, et al. A phase 1/2, dose-escalation trial of deferasirox for the treatment of iron overload in HFE-related hereditary haemochromatosis. Hepatology 2010;52:1671-779.
Phelps G, Chapman I, Hall P, et al. Prevalence of genetic haemochromatosis among diabetic patients. Lancet 1989;2:233-4.
Pietrangelo A, Montosi G, Totaro A. Hereditary haemochromatosis in adults without pathogenic mutations in the haemochromatosis gene. N Engl J Med 1999;341:725-32.
Pietrangelo A. Physiology of iron transport and the haemochromatosis gene. Am J Physiol Gastrointest Liver Physiol 2000;282:G403-14.
Pietrangelo A. Hereditary haemochromatosis -a new look at an old disease. N Engl J Med 2004;350:2383-97.
Pietrangelo A. The ferroportin disease. Blood Cells Mol Dis 2004;32:131-8.
Pietrangelo A. Ferroportin disease: pathogenesis, diagnosis and treatment. Haematologica 2017;102:1972-84.
Piga A, Galanello R, Forni GL, et al. Randomised phase II trial of deferasirox (Exjade, ICL670), a once-daily, orally-administered iron chelator, in comparison to deferoxamine in thalassaemia patients with transfusional iron overload. Haematologica 2006;91:873-80.
Piga A, Longo F, Duca L, et al. High nontransferrin bound iron levels and heart disease in thalassaemia major. Am J Hematol 2009;84:29-33.
Piperno A, Roetto A, Mariani R, et al. Homozygosity for transferrin receptor-2 Y250X mutation induces early iron overload. Haematologica 2004; 89:359-60.
Piroth L, Meyer P, Bielefeld P, Besancenot JF. Yersinia bacteraemia and iron overload. Rev Med Interne 1997;18:932-8.
Pootrakul P, Breuer W, Sametband M, et al. Labile plasma iron (LPI) as an indicator of chelatable plasma redox activity in iron overloaded beta-thalassaemia/HbE patients treated with an oral chelator. Blood 2004;104:1504-10.
Porter JB Practical management of iron overload. Br J Haematol 2001;115:239-52.
Powell LW, Mortimer R, Harris OD. Cirrhosis of the liver: A comparative study of the four major aetiological groups. Med J Aust 1971;58:1941-50.
Radojska S, Hengeoz O, Gathof B. Hereditary haemochromatosis patients/ carriers as blood donors – a single center experience. Transfus Med Hemother 2011;38:32.
Rahier J, Loozen S, Goebbels RM, et al. The haemochromatosis human pancreas: a quantitative immunochemical and ultrastructural study. Diabetologia 1987;30:5-12.
Recklinghausen von FD. Über Hämochromatose. Berl Klin Wochenschr 1898;26:925.
Rivard SR, Lanzara C, Grimard D, et al. Autosomal dominant reticuloendothelial iron overload (HFE type 4) due to a new missense mutation in the FERROPORTIN 1 gene (SLC11A3) in a large French-Canadian family. Haematologica 2003;88:824-6.
Rombout-Sestrienkova E, Nieman FH, et al. Erythrocytapheresis versus phlebotomy in the initial treatment of HFE haemochromatosis patients: results from a randomised trial. Transfusion 2012;52:470-7.
Rombout-Sestrienkova E, Winkens B, et al. Erythrocytapheresis versus phlebotomy in the maintenance treatment of HFE haemochromatosis patients: results from a randomised crossover trial. Transfusion. 2016;56:261-70.
Roetto A, Totaro A, Cazzola M, et al. Juvenile haemochromatosis locus maps to chromosome 1q. Am J Hum Genet 1999;64:1388-93.
Roetto A, Totaro A, Piperno A, et al. New mutations inactivating transferrin receptor 2 in haemochromatosis type 3. Blood 2001;97:2555-60.
Sackett DL, Rosenberg MC, Gray JA, Haynes RB, Richardson WS. Evidence based medicine: what it is and what it isn’t. BMJ 1996;312:71-2.
Sanchez AM, Schreiber GB, Bethel J, et al. Prevalence, donation practices, and risk assessment of blood donors with haemochromatosis. J Am Med Assoc 2001;286:1475-81.
Schuhmacher HR. Haemochromatosis and arthritis. Arthr Rheum 1964;7:41-50.
Senba M, Nakamura T, Itakura H. Relationships among iron accumulation, cirrhosis, and hepatitis B virus infection in Bantu siderosis. Ann Soc Belg Med Trop 1989;69:77-8.
Sheldon JH. Haemochromatosis. Oxford University Press, 1935, London.
Short EM, Winkle RA, Billingham ME. Myocardial involvment in idiopathic haemochromatosis. Am J Med 1979;70:1275-9.
Singh BM, Grunewald RA, Press M, et al. Prevalence of haemochromatosis amongst patients with diabetes mellitus. Diabet Med 1992;9:730-1.
Simon M, Pawlotsky Y, Bourel M, et al. Hemochromatose idiopathique. Maladie associee a l’antigen HLA-A3? Nouv Press Med 1075;4:1432.
Simon M, Bourel M, Genetet B. Idiopathic haemochromatosis: demonstration of recessive transmission and early detection by family HLA typing. N Engl J Med 1977;297:1017-21.
Summers KM, Halliday JW, Powell LW. Identification of homozygous haemochromatosis subjects by measurement of hepatic iron index. Hepatology 1990;12:20-5.
Sundic T, Hervig T, Hannisdal S, et al. Erythrocytapheresis compared with whole blood phlebotomy for the treatment of hereditary haemochromatosis. Blood Transfus 2014;12:S84-9.
Swart de L, Hendriks JC, van der Vorm LN, et al. Second International Round Robin for the quantification of serum Non-Transferrin-Bound Iron and Labile Plasma Iron in patients with iron-overload disorders. Haematologica 2015:133983.
Taranath RP, Dion C, Bhandari A, Liu DY. Rusfertide analog-PN23114 as a hepcidin mimetic provides efficacy benefits in conjunction with phlebotomy in mouse model for hereditary haemochromatosis. Blood 2022;14: S5345.
Tavill AS. Diagnosis and management of haemochromatosis. Hepatology 2001;33:1321-8.
Telfer PT, Prestcott E, Holden S, et al. Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major. Br J Haematol 2000;110:971-7.
Toll A, Celis R, Ozalla MD, et al. The prevalence of HFE C282Y gene mutation is increased in Spanish patients with porphyria cutanea tarda without hepatitis C virus infection. J Eur Acad Dermatol Venereol 2006;20:1201-6.
Troisier M. Diabete sucre. Bull Soc Anatom (Paris) 1871;16:231-5.
Trosseau A. Glucosurie: Diabete sucre. Bull Soc Anatom (Paris) 1865;2:663.
U.S. Department of Health and Human Services, Food and Drug Administration, Center for Biologics Evaluation and Research (CBER). Guidance for industry Variances for Blood Collection from Individuals with Hereditary Haemochromatosis 2001. Available from: http://www.fda.gov/cber/guidelines.htm.
U.S. Preventive Services Task Force. Screening for Haemochromatosis: Recommendation Statement. Am Fam Phys 2007;75:11-21.
Vaiopoulos G, Papanikolaou G, Politou M, et al. Arthropathy in juvenile haemochromatosis. Arthritis Rheum 2003;48:227-30.
Vanclooster A, van Deursen C, Jaspers R, Cassiman D, Koek G. Proton pump inhibitors decrease phlebotomy need in HFE haemochromatosis: double-blind randomised placebo-controlled trial. Gastroenterology 2017;153:678-80.
Vichinsky E, Fischer R, Pakbaz Z, et al. Satisfaction and convenience of chelation therapy in patients with sickle cell disease (SCD): comparison between deferasirox (Exjade®, ICL670) and deferoxamine (DFO). Blood 2005;106:A2334.
Vulpe CD, Kuo YM, Murphy TL, et al. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet 1999;21:195-9.
Wallace DF, Clark RM, Harley HA, et al. Autosomal dominant iron overload due to a novel mutation of ferroportin1 associated with parenchymal iron loading and cirrhosis. J Hepatol 2004;40:710-3.
Wallace DF, Subramaniam VN. Non-HFE haemochromatosis. World J Gastroenterol 2007;13:4690-8
Walker EM, Walker SM. Review: Effects of iron overload on the immune system. Ann Clin Lab Sci 2000;30:354-65.
Yaouanq JM. Diabetes and haemochromatosis: current concepts, management and prevention. Diabete et Metabolisme. (Paris) 1995;21:319-29.
Ye Q, Qian BX, Yin WL, Wang FM, Han T. Association between the HFE C282Y, H63D polymorphisms and the risks of non-alcoholic fatty liver disease, liver cirrhosis and hepatocellular carcinoma: An updated systematic review and meta-analysis of 5, 758 cases and 14, 741 controls. PLoS One 2016;11:e0163423.
Zhang AS, Xiong S, Tsukamoto H, et al. Localisation of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes. Blood 2004;103:1509-14.
Zurlo MG, De Stefano P, Borgna-Pignatti C, et al. Survival and causes of death in thalassaemia major. Lancet 1989;2:27-30.