
10. Genetic liver diseases
10.4 Alpha-1 antitrypsin deficiency
Michelle Walkenhaus, Pavel Strnad
Abstract
Alpha-1 antitrypsin (AAT) deficiency (AATD) is caused by a mutation in the SERPINA1 gene, which encodes the protease inhibitor alpha1 antitrypsin. This leads to AAT retention in the hepatocytes. As a result, a proteotoxic stress reaction occurs in the liver and predisposes to liver fibrosis/cirrhosis (gain-of-function). Less AAT reaches the lungs, which can lead to pulmonary emphysema or COPD (loss-of-function). The homozygous Pi*ZZ genotype causes severe AATD and may result in paediatric and adult liver diseases. The heterozygous Pi*MZ genotype is a risk factor for the development of lung and liver diseases. The progression of lung disease can be slowed down by AAT substitution therapy and a phase II clinical study suggests a therapeutic benefit of AAT silencing via siRNA for AATD-related liver disease. This chapter summarises the pathomechanisms, diagnostic approach, clinical consequences as well as recommendations for clinical management of the subjects with AATD.
Background
Alpha-1 antitrypsin deficiency (AATD) is a common inherited disorder that is potentially life-threatening due to the associated lung and liver damage (Greene 2016). In the ICD-10 nomenclature, it is reflected by an unspecific code E88.0 and because of that, Orphanet nomenclature of rare diseases (ORPHA) code 60 represents a more specific way to identify this condition. Alternatively, ICD modifications such as E88.0A code that is used in some European countries or the upcoming ICD-11 system offer a better assignment (Picker 2023). AATD arises due to mutations in the SERPINA-1 gene, located on chromosome 14. It encodes the protein alpha-1 antitrypsin (AAT), a protease inhibitor that is mainly produced in hepatocytes. After being secreted from the hepatocytes into the bloodstream, AAT inhibits a variety of proteases such as neutrophil elastase or proteinase 3 thereby preventing an undesired tissue digestion (Greene 2016). The liver produces up to 34 mg of AAT per kilogram of body weight per day. It results in serum levels of 0.9 to 1.75 mg per millilitre in healthy individuals. AAT is a stress-inducible protein and consequently, this level can be doubled during infections. In individuals with AATD, AAT production/secretion is impaired. This fact results in diminished serum AAT levels and a less pronounced increase during infections (Strnad 2020).
Most individuals have two wild type alleles of the SERPINA-1 gene, labeled as Pi*MM genotype. More than 100 AAT variants have been described and they are subdivided into groups based on the migration of the resulting protein in the electric field. The wild type allele Pi*M indicates a medium velocity, while Pi*F, Pi*S and Pi*Z refer to fast, slow and very slow movement, respectively (Wedzicha 2017). The most clinically relevant variants are Pi*Z (glutamate-to-lysine substitution at position 342) and Pi*S (glutamate-to-valine substitution at position 264) (Ferrarotti 2014). Depending on the amount of affected alleles, they cause the genotypes Pi*MZ/Pi*ZZ (heterozygous/ homozygous Pi*Z mutation) and Pi*MS/SS (heterozygous Pi*S mutation/ homozygous Pi*S mutation), while a combined presence of Pi*Z and Pi*S variant is termed Pi*SZ (Ferrarotti 2014, Strnad 2020).
Given the AATD pathomechanism described above, measurement of the AAT serum level is the first step in the detection of this condition. When decreased levels are seen, a genetic analysis and/or protein phenotyping is usually performed. While a cut-off of <50mg/dL can be used to identify individuals with severe AATD genotypes, there is no level reliably identifying subjects with mild genotypes such as Pi*MZ although a level of 110mg/dL has been advocated by some researchers (Ferrarotti 2012, Miravitlles 2017). The genotyping can be performed via a cheek swab or a blood draw with DNA isolation. A routine diagnosis typically consists of a PCR for Pi*Z/Pi*S or a multiplex-PCR that distinguishes ~20 most common variants. If this approach does not yield a definitive diagnosis or the detected genotype does not match with serum AAT levels, a sequencing is typically performed (Strange 2006, Strnad 2020). As an alternative to genotyping, AAT phenotyping takes advantage of different migration in the electric field described above and because of that provides an assignment to subclasses rather than a final genetic diagnosis (Bornhorst 2013). The major advantage of that approach is that it is often faster than the definitive genetic diagnosis.
While the liver and lung affection are the major AATD-associated conditions, the underlying mechanism differs. In the former case, the abnormal AAT that cannot be released into the bloodstream, polymerises and causes a toxic “gain-of-function” injury (Clark 2018, Schneider 2020). The hepatocellular retention of the heavily glycosylated AAT leads to an emergence of PAS-D positive globules that are the histological hallmark of this condition. In contrast, the lung suffers from the "loss-of-function" phenotype caused by the lower amount of AAT in the systemic circulation and consequently the lung. This results in an impaired inhibition of the target proteases and an increased degradation of the alveolar septa. This leads to panlobular emphysema and chronic obstructive pulmonary disease (COPD) that are the prevailing AATD-specific cause of death (Greene 2016, Janciauskiene 2018, Janciauskiene 2011). Notably, additional events such as inflammatory reaction caused by the polymerised AAT might also contribute to the lung damage but their exact contribution remains to be clarified.
The different mechanistic cause of lung and liver affection becomes important when counseling subjects with the rare, so called "null mutations" (Pi*Q0), i.e. mutations that do not display any serum AAT. Because of the lack of the AAT, the affected individuals are highly predisposed to lung damage while their liver-related risks differ, depending on whether or not they produce any hepatic AAT (Ferrarotti 2014).
Besides being an anti-protease, AAT is also an important immunomodulatory protein and consequently, AATD subjects are susceptible to immunologic disorders such as antineutrophil cytoplasmic antibody (ANCA)– associated vasculitis or panniculitis, a disorder characterised by subcutaneous inflammation. Accordingly, current guideline suggests that individuals with the latter two conditions, COPD, liver disease of unknown origin, asthma not responsive to the treatment, bronchiectasis as well as first-degree relatives of AAT subjects should be tested for this condition. Given that AATD can be both liver disease-causing and –modifying, we advocate for AATD testing in all subjects where the result might be of clinical relevance.
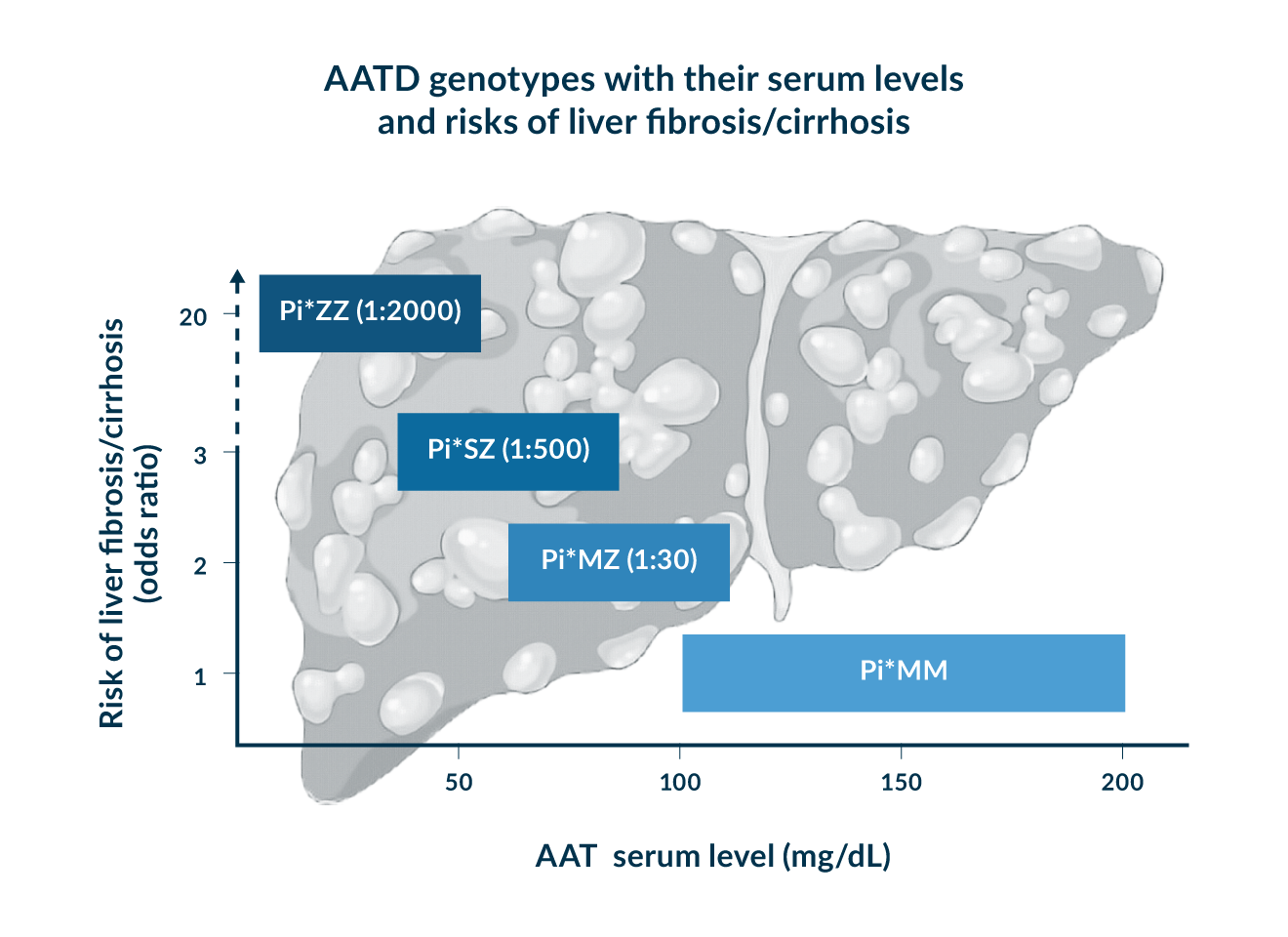 Figure 1. Characteristic AATD genotypes, their prevalence, AAT serum levels and corresponding risk of adult liver fibrosis/cirrhosis.
Figure 1. Characteristic AATD genotypes, their prevalence, AAT serum levels and corresponding risk of adult liver fibrosis/cirrhosis.
Pi*ZZ
The genotype Pi*ZZ is responsible for >95% of severe AATD cases and occurs at a frequency of 1:2000 in subjects of European ancestry (Blanco 2017). Patients with the corresponding homozygous Z mutation, have a markedly decreased serum AAT level (mostly <50 mg/dL AAT serum level) and a marked susceptibility to liver and lung damage (Hamesch 2019, Strnad 2020, Teckman 2013).
Adult liver
Pi*Z mutation does not affect the production of AAT protein in the hepatocytes but leads to protein misfolding and an 85% reduction in secreted AAT. As a result, the protein is retained in the endoplasmic reticulum (ER) (Lomas 1992, Teckman 2017). About 70% of it can be degraded while the rest forms polymers. The degradation occurs with the help of autophagy and ER-associated degradation (ERAD) (Teckman 2000, Christianson 2008). ERAD is responsible for removal of smaller species which autophagy can degrade even larger aggregates. These can be visualised as purple inclusion bodies in the periodic Acid-Schiff staining and are resistant to diastase treatment (PAS-D stain) that is used to digest glycogen. Immunohistochemistry using anti-Pi*Z antibodies is even more sensitive but is rarely used in the clinical routine (Strnad 2013). On the other hand, AAT aggregates are rather indistinct in the hematoxylin and eosin staining. The misfolding and polymerisation of the AAT leads to a proteotoxic “gain-of-function” stress, which promotes the development of liver fibrosis/cirrhosis (Clark 2018, Schneider 2020). Accordingly, Pi*ZZ subjects display higher liver transaminases (AST, ALT) as well as GGT values (Fromme 2022b). However, elevated AST/ALT levels are seen only in 10-15% of Pi*ZZ subjects and repeatedly elevated levels should therefore trigger thorough evaluation to exclude potential co-morbidities.
Despite the often normal liver enzyme levels, Pi*ZZ patients show a 20-fold higher risk of developing liver fibrosis/liver cirrhosis compared to the normal population and the life-long risk of cirrhosis of about 10%. These data are supported by several independent sources. Among them, the United Kingdom Biobank (UKB), a population-based cohort of ~500, 000 individuals who all received AATD genotyping, assessed the presence of advanced liver fibrosis based on ICD codes documented during inpatient visits (Fromme 2022a). The EASL AATD consortium collected almost 600 Pi*ZZ subjects, who were examined by transient elastography-based liver stiffness measurement (LSM) as well as other non-invasive liver fibrosis surrogates such as AST-to-platelet ratio (APRI) or Fib-4 index (Schneider 2020, Hamesch 2019). Another study compared the need for liver transplantation and came to similar conclusions. (Adam 2012, Fromme 2022a). Finally, a cohort from North America subjected 94 Pi*ZZ individuals to a liver biopsy and found that 35% of them had significant liver fibrosis defined as a fibrosis grade of at least 2 on a 0-4 scale (Clark 2018). This study as well as a report from the EASL AATD consortium demonstrated the usefulness of non-invasive fibrosis parameters (particularly LSM) to reflect the histological amount of liver fibrosis. Similar to other etiologies, LSM was particularly useful to recognise advanced fibrosis while its performance in intermediate fibrosis stages was less impressive (Clark 2018). Clark et al also revealed that increased fibrosis stage associates with higher amount of AAT inclusions. However, further studies are needed whether inclusion load really increases during fibrosis progression or whether subjects with increased AAT accumulation are more susceptible to the fibrosis development.
In contrast to data on liver fibrosis, there are only limited data on susceptibility to liver tumours (Fromme 2022a, Hamesch 2019, Schneider 2020). The UKB analyses showed an odds ratio of > 40, while another study reported a ~20x increased risk (Hiller 2022). However, it remains unclear whether tumours develop only at advanced fibrosis stage, as seen in most liver disease etiologies or can also arise earlier. Because of that, we advocate for regular liver tumour screening in Pi*ZZ subjects with at least an intermediate liver fibrosis stage (Fromme 2022a).
Given the above described risks, a hepatological assessment of Pi*ZZ subjects is warranted. For an initial evaluation, we recommend a combination of liver enzyme levels and LSM. A LSM≥7.1 kPa suggests a fibrosis of grade 2 or higher, while LSM≥10 kPa points towards an advanced liver fibrosis. Ideally, LSM values are in accordance with at least one widely available non-invasive liver fibrosis surrogate such as APRI or Fib-4. In discrepant cases, magnetic resonance elastography can be used as an additional methods although the data on its usefulness in AATD are highly limited (Kim 2016). While liver biopsy is not needed to establish the diagnosis, it might be useful in case of recurrently elevated liver enzymes to assess co-morbidities or to clarify the extent of fibrosis in unclear cases. In addition to the LSM, transient elastography can be used to quantify steatosis of the liver via assessment of controlled attenuation parameters (CAP). Notably, Pi*ZZ individuals display increased CAP values (Hamesch 2019).
A longitudinal examination of Pi*ZZ subjects should be based on results of the initial evaluation. Given their markedly increased risks, we advocate for measurement of the inexpensive liver enzymes once or twice a year. However shorter intervals might be needed in subjects with increased values or advanced fibrosis. We recommend an ultrasound of the liver once a year to exclude tumours, at least in subjects with intermediate fibrosis and twice a year in individuals with advanced liver fibrosis. If the baseline transient elastography and the liver values are unremarkable, a further transient elastography can occur in about 3 years. Patients with decompensated liver cirrhosis should be considered for liver transplantation and the evaluation should include a careful examination of the lung status.
Paediatric liver
While adult AATD-associated liver disease (AATD-LD) typically manifests in/after 40 years of age, Pi*ZZ genotype also causes paediatric liver damage that often manifests in form of neonatal cholestasis (Ruiz 2019). It is characterised by prolonged cholestatic jaundice that is accompanied by increased serum enzyme levels. It can lead to failure to thrive and hepatosplenomegaly and progress to liver failure with ascites and coagulopathy (Feldman 2013).
The highest level of evidence about paediatric AAT-LD stems from the Swedish Newborn Screening programme, which screened a cohort of 200, 000 newborns and identified 120 Pi*ZZs (Mostafavi 2019, Sveger 1976, Sveger 1995). 12% of them displayed prolonged jaundice (Sveger 1976). Over half of the children with the Pi*ZZ genotype showed abnormal liver enzymes and these values often normalised in the next months/years (Sveger 1995). Less than 3% of the Pi*ZZ newborns developed an end-stage liver disease. At the age of 18 years, only 12% of the Pi*ZZ subjects had elevated liver enzyme levels (Ruiz 2019, Fromme 2019, Sveger 1995). The programme also systematically evaluated the psychological burden of the disease and layed out both advantages of disadvantages of neonatal screening for AATD.
While some Pi*ZZ infants/children develop typical symptoms of decompensated liver cirrhosis, such as a distended abdomen or gastrointestinal (GI) bleeding from oesophageal varices, many remain asymptomatic even in case of portal hypertension and are discovered through checkups or biochemical screenings. These included altered liver enzymes, thrombocytopenia or splenomegaly. Some have a history of prolonged jaundice as a newborn, while others are first noticed in screening procedures later on. In general, children with symptoms of chronic liver disease, hepatosplenomegaly, unexplained jaundice or abnormal liver enzymes should be tested for AATD. Similar to adults, liver biopsy is not needed to diagnose the disease but might be useful in unclear cases (Feldman 2013).
Similar to adults, a management of paediatric AATD-LD is based on the rate of liver affection. Given that the course of the disease can be very variable and rapid decompensations have been described in those with advanced liver fibrosis, a careful monitoring by a paediatric liver transplantation centre is recommended. Those with signs of portal hypertension and failure to thrive should be considered for liver transplantation. Since cholestatic liver disease impairs the absorption of fat-soluble vitamins, an evaluation of vitamin deficiencies and their supplementation might be needed. Further liver damage from alcohol or a steatosis should be avoided and a healthy lifestyle promoted (Feldman 2013).
Although no specific treatment of paediatric AATD-LD exists, ursodeoxycholic acid (UDCA) is frequently administered, although the data are based only on one open-label trial. In the latter, 42 Pi*ZZ children with the Pi*ZZ genotype were assessed over the course of several years. They all received UDCA and were clinically and biochemically examined at least once a year. UDCA improved liver test results in some, but no benefit was seen in those with pre-existing severe liver damage (Lykavieris 2008).
Given the marked liver- and lung-related risks associated with the Pi*ZZ genotype, a systematic transitional programme for adolescents is warranted. The topics should include the awareness about disease, the promotion of healthy lifestyle, avoidance of smoking, dust exposition or excessive alcohol consumption. In addition, the importance of regular lung and liver monitoring as well as the usefulness of vaccinations should be addressed.
Lung
In contrast to liver injury, proteolytic lung damage is caused by a “loss-of-function” mechanism. The strongly reduced AAT levels in the blood result in an insufficient inhibition of the proteases, particularly neutrophil elastase (Janciauskiene 2011). This leads to increased degradation of the alveolar septa, an elevated mucin production and secretion as well as activation of several proinflammatory downstream targets (Strnad 2020). In addition to that, AAT is a potent anti-inflammatory agent and its lack promotes neutrophil chemotaxis, degranulation and apoptosis (Strnad 2020). To make things worse, the polymerised, misfolded AAT also constitutes a proinflammatory agent and a potent neutrophil chemoattractant (Mahadeva 2005). Smoking oxidises and thereby inactivates AAT, but also induces its polymerisation (Alam 2011). Collectively, the effects lead to panlobular emphysema or chronic obstructive pulmonary disease (COPD) that often becomes symptomatic in fourth or fifths decade and develops particularly rapidly, especially in smokers. The lung damage is the most common specific cause of death in Pi*ZZ patients (Tanash 2017). Therefore, smoking Pi*ZZ subjects should be offered a counselling to help them with smoking cessation. Notably, early intervention reduced the frequency of smoking and mortality (Piitulainen 2002).
The monitoring of Pi*ZZ subjects with lung disease involves spirometry assessing the mean forced expiratory volume in 1 second (FEV1) a measurement of diffusing capacity of the lung for carbon monoxide (DLCO), a 6-minute walk test and a quality-of-life questionnaire (Miravitlles 2017). The frequency of check-ups depends on the degree of impairment. At the beginning, a biannual testing is appropriate, while a once-a-year evaluation is often sufficient later on. COPD guidelines can be used as a guide (Strnad 2020). Notably, neither FEV1 nor DLCO reflect the full lung phenotype and lung density assessment via computed tomography has been advocated as a better tool, however, long-term studies analysing its predictive value are lacking (McElvaney 2017).
The treatment of Pi*ZZ-associated lung disease follows the recommendations for COPD. Additionally, the loss of lung density can be slowed down by weekly intravenous administration of purified human AAT (Greene 2016, Strnad 2020). Notably, AAT augmentation does not seem to affect COPD exacerbations nor FEV1 (McElvaney 2017, Chapman 2015). AAT augmentation infusion is recommended for non-smoking patients with severe AATD with diminished FEV1, frequent exacerbation or a rapid decline in lung function. However, AAT augmentation is not reimbursed in all countries and is not recommended for subjects with mild AATD such as Pi*MZ nor for patients without airflow obstruction (Sandhaus 2016, Miravitlles 2017).
In addition to the above described measures, the management of PI*ZZ patients should include a prevention and aggressive treatment of respiratory infections. For the former, regular vaccinations against influenza, SARS-CoV2 and pneumococci are recommended (Miravitlles 2017). The subjects with chronic respiratory failure benefit from long-term oxygen therapy. Lung transplantation, surgical lung volume reduction and endobronchial valve placement might be helpful in patients with advanced emphysema, although the latter two are controversial and require a careful selection (Miravitlles 2017).
Others
AATD constitutes a systemic disease and in addition to lung and liver, it may also affect other organs. In particular, about 1% of Pi*ZZ subjects develop a neutrophilic panniculitis (Blanco 2016), a painful inflammation of the subcutaneous fat tissue characterised by nodular or ulcerating skin lesions. Some patients also report a discharge of a yellow exudate (Franciosi 2022). The upper and lower extremities are most commonly affected, but the back, abdomen or face can also be involved. The disease can occur gradually or intermittently in episodes. It predominantly affects women and the typical age at first presentation is 36 years (Franciosiz 2015, Elsensohn 2015). The first-line treatment is dapsone, whereas AAT augmentation therapy is used in refractory cases. Because of the option for a specific treatment, it is important to identify AATD as the underlying cause, especially since panniculitis associated with AATD is often resistant to treatment with glucocorticoids (Franciosi 2022).
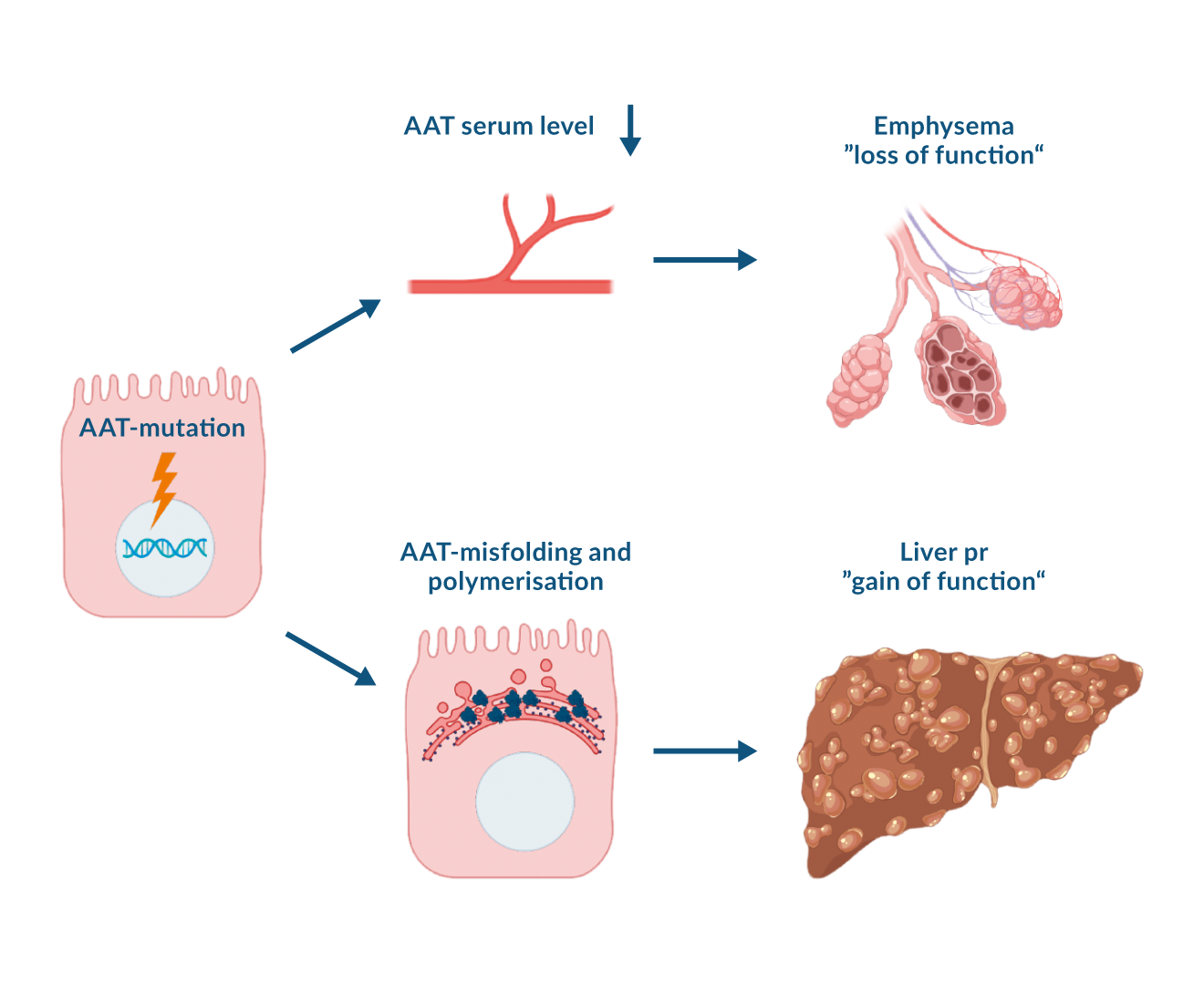 Figure 2. Pathophysiology of liver and lung damage in alpha-1 antitrypsin deficiency. Alpha-1 antitrypsin (AAT) mutation causes AAT accumulation and a consecutive proteotoxic „gain-of-function“ stress in the liver. The impaired AAT secretion leads to a „loss-of-function“ lung injury due to a reduced inhibition of proteases.
Figure 2. Pathophysiology of liver and lung damage in alpha-1 antitrypsin deficiency. Alpha-1 antitrypsin (AAT) mutation causes AAT accumulation and a consecutive proteotoxic „gain-of-function“ stress in the liver. The impaired AAT secretion leads to a „loss-of-function“ lung injury due to a reduced inhibition of proteases.
Pi*MZ
The heterozygous genotype Pi*MZ is found in about 1:30 Caucasians. These individuals have normal to slightly decreased serum AAT levels (typically 70-100 mg/dL). Many of them are discovered during family screening of Pi*ZZ relatives.
Liver
In this genotype, the presence of a Z mutation is partially compensated by the remaining wild-type allele. This means, that more AAT is transported into the bloodstream and less misfolded AAT remains in the ER. This results in less polymerisation seen in the liver biopsies. For example, in a large study, only 40 % of Pi*MZ patients harbored AAT inclusion bodies in the PAS-D staining, while immunohistochemistry detected AAT polymers in 63 % of them (Fu 2017, Schneider 2020). In line with that, in a population-based UKB cohort, individuals with Pi*MZ showed only a slightly increased odds ratio of 1.7 for liver fibrosis/ liver cirrhosis as well as liver mortality and no increased risk for liver cancer (Fromme 2022a, Hamesch 2019, Schneider 2020). Therefore, Pi*MZ genotype is considered a risk factor rather than a disease-causing agent and a second hit is typically needed to develop a disease (Schneider 2020). For example, the genetically determined 1.7-fold risk of liver-related mortality is amplified by established factors such as obesity, diabetes mellitus, male gender and age ≥ 50 years (Schneider 2020). Among them, obesity is well-known to increase the profibrogenic effects of several other genetic variants (Williams 2014, Stender 2017), while diabetes was suggested to promote proteotoxic cell stress, oxidative stress and lipolysis (Lebeaupin 2018, Petersen 2018). Several studies have shown that the Pi*MZ genotype increases susceptibility to liver fibrosis in metabolic disorders such as ALD/NAFLD. The odds ratio for NAFLD-related cirrhosis in subjects with Pi*MZ genotype was 2-7 times higher than in non-carriers and similar magnitude was reported for ALD (Strnad 2019, Abul-Husn 2018, Cacciottolo 2014). In contrast, the interaction between Pi*MZ genotype and viral hepatitis or haemochromatosis remains to be conclusively clarified (Guldiken 2019, Regev 2006, Kuscuoglu 2021), while the worsening impact on cystic fibrosis-related liver disease is unequivocal (Strnad 2020) and confers an odds ratio of ~5 (Ruiz 2023). Notably, cystic fibrosis constitutes a cholestatic disease and the detrimental impact of Pi*MZ phenotype on proper bile secretion is also supported by their predisposition to gallstone formation (Ferkingstad 2018).
In line with the above described considerations, liver enzyme levels are typically within normal range in Pi*MZ subjects although the mean ALT values in the UKB cohorts were significantly elevated compared to non-carriers (Fromme 2022a). It is worthwhile to note that up to 10% of liver transplant candidates carry a Pi*MZ genotype and that Pi*MZ subjects with liver cirrhosis decompensate faster than non-carriers (Ruiz 2023). Therefore, a liver check-up should be offered to Pi*MZ subjects with established risk factors and/or elevated liver enzymes, while there are no sufficient data to recommend a systematic hepatologic management of asymptomatic Pi*MZ individuals.
Lung
Similar to liver disease, the Pi*MZ genotype alone does not constitute a dramatic risk factor for the development of lung disease. In fact, population studies did not identify an elevated predisposition on a population level (Strnad 2020), while several reports demonstrated an increased susceptibility in presence of additional risk factors or a permissive genetic background (Molloy 2014).
For example, the COPDGene study investigated the lung function of current and ex-smokers with at least 10 pack-years using a cross-sectional observational cohort. It was established that Pi*MZ patients have significantly lower FEV1 values and more radiological emphysema than non-carriers. Notably, the phenotype was shown across all races (Foreman 2017).
Other studies showed that the significant difference between Pi*MZ and Pi*MM subjects is not observed in never smokers and particularly pronounced in continuous smokers. The odds ratio was the highest by forever-smokers at 10.65 (Molloy 2014). These data should be used to discourage Pi*MZ subjects from smoking and to include them in corresponding preventative programmes. While not every Pi*MZ person needs regular pulmonological examinations, these should be considered in those with additional risk factors such as smoking or pre-existing COPD or asthma.
Rare associations
Milder genotypes such as the Pi*MZ do not seem to per se trigger a development of liver disease in children, but can contribute to such a disease in presence of additional risk factors such as cystic fibrosis (Ruiz 2019). However, partly due to their abundance, several disorders were shown to be associated with Pi*MZ genotype (Strnad 2020).
For example, Pi*MZ subjects are susceptible to gallstone formation with an odds ratio of 1.3 for both Pi*ZZ and the Pi*MZ genotype (Ferkingstad 2018). Pi*MZ individuals also harbor a 2.9times elevated risk of ANCA-associated vasculitis (Merkel 2017) with a clear data for both myeloperoxidase-reactive ANCA with perinuclear staining (p-ANCA) and proteinase 3-reactive ANCA with cytoplasmic staining (c-ANCA) subtypes. This overrepresentation was detected not only in individuals with a Pi*Z mutation, but also in those with a Pi*S mutation. The pathomechanism seems to be a loss-of-function of AAT leading to an imbalanced protease activity (Rahmattulla 2016).
Less common genotypes
While >100 genotypes have been described (Strnad 2020), Pi*MZ and Pi*ZZ are the clinically by far most relevant ones. Additionally, Pi*SZ genotype is also relatively common (ca. 1:500-1:1000 in Caucasians) and displays intermediate AAT serum levels (ca. 40-80 mg/dL) (Fromme 2022a). Susceptibility to lung and liver diseases is moderately increased, but seems to be closer to Pi*MZ than Pi*ZZ subjects (Blanco 2017, Fromme 2022a). For example, Pi*SZ individuals have a 3-fold higher risk of liver fibrosis or cirrhosis, but a 7-fold higher risk of liver cancer (Fromme 2022a). Therefore, regular ultrasound examinations should be performed when advanced liver fibrosis is present. The risk factors that promote the development of liver fibrosis seem to be the same (i.e. male gender, ≥ 50 years, metabolic syndrome) as reported above for Pi*MZ subjects (Fromme 2022a). Along the same line, Pi*SZs should be discouraged from smoking. While non-smoking Pi*SZ individuals with lung disease are offered augmentation therapy in some countries, but not in others (Cazzola 2020).
The genotype Pi*SS has a comparable prevalence as Pi*SZ but does not seem to be a relevant risk factor for the development of lung and liver diseases (Fromme 2022a). Paediatric liver diseases do not typically develop in the less severe genotypes such as Pi*SZ and Pi*SS, but these genotypes may play a role in the presence of other risk factors (Ruiz 2019). In the even less frequent genotypes, the risk of lung disease seems to be indirectly proportional to the serum AAT level, while the liver related risk also depends on the underlying biology as described above for the Pi*Q0 variants (Strnad 2020).
Therapeutic approaches for AATD-related liver disease
The current treatment options in AATD are limited to lung/liver transplantation and the augmentation therapy. Liver transplantation is associated with a good prognosis in carefully selected patients (Carey 2013, Clark 2017) and since the majority of human AAT is produced in the liver, results in a normalisation of serum AAT levels. While this fact should protect from progression of lung disease, the current data supporting this consideration are limited (Carey 2013). Similarly, AATD candidates with sever lung damage were demonstrated to benefit from lung transplantation (Tanash 2011), although they might have a faster FEV1 decline than COPD patients without AATD (Banga 2014). Although experimental data suggest a beneficial impact of AAT augmentation after lung transplantation (Iskender 2016), lung-transplanted AATD subjects are not regularly augmented.
In line with the pathomechanism of AATD-associated lung disease described above, lung-directed candidates focus on AAT supplementation or on inhibition of its target protein. With regard to the former, an inhaled AAT was demonstrated to be safe in a phase 2 clinical trial (Stolk 2019) and its efficacy is currently studied in a phase 3 trial. While the current augmentation therapy relies on purified human protein, inhibrix-101 constitutes a recombinant AAT form with a longer half-life that is studied in early phase clinical trials (Kuhn 2023). Alvelestat is an oral inhibitor of neutrophil elastase and in a phase 2 trial, its administration at higher dose was associated with decreased neutrophil elastase activity (Stockley 2023).
On the other hand, liver-targeted treatments aim to decrease the production of the misfolded protein or improve its secretion from the liver. The former strategy relies primarily on hepatocyte-targeted small interfering RNAs (siRNAs) fazirsiran or belcesiran. In a phase 2 open-label study, fazirsiran reduced hepatic AAT accumulation as well as the levels of Z-AAT in serum by 85% and this reduction was associated with improved liver enzyme levels as well as decreased liver fibrosis (Strnad 2020). While fazirsiran (also labeled as TAK-999) is currently tested in a placebo-controlled phase 3 trial, belcesiran is currently in a phase 2 trials and no efficacy data for AATD subjects exist. Several other strategies aiming to reduce hepatic AAT production such as CRISPR/Cas approach, gene or RNA editing are at a pre-clinical or early clinical level (Ruiz 2023, Strnad 2023). As a complementary approach, the so called folding correctors aim to promote AAT folding and the consecutive secretion from the liver. In an ideal case, this would improve both lung and liver disease, however, the candidates tested up to date led to only minor increases in serum AAT levels and in part also displayed si gnificant adverse effects (Ruiz 2023). Despite that, AATD constitutes an important model disease promoting the development of several modern therapies, mainly in the area of genetic medicine.
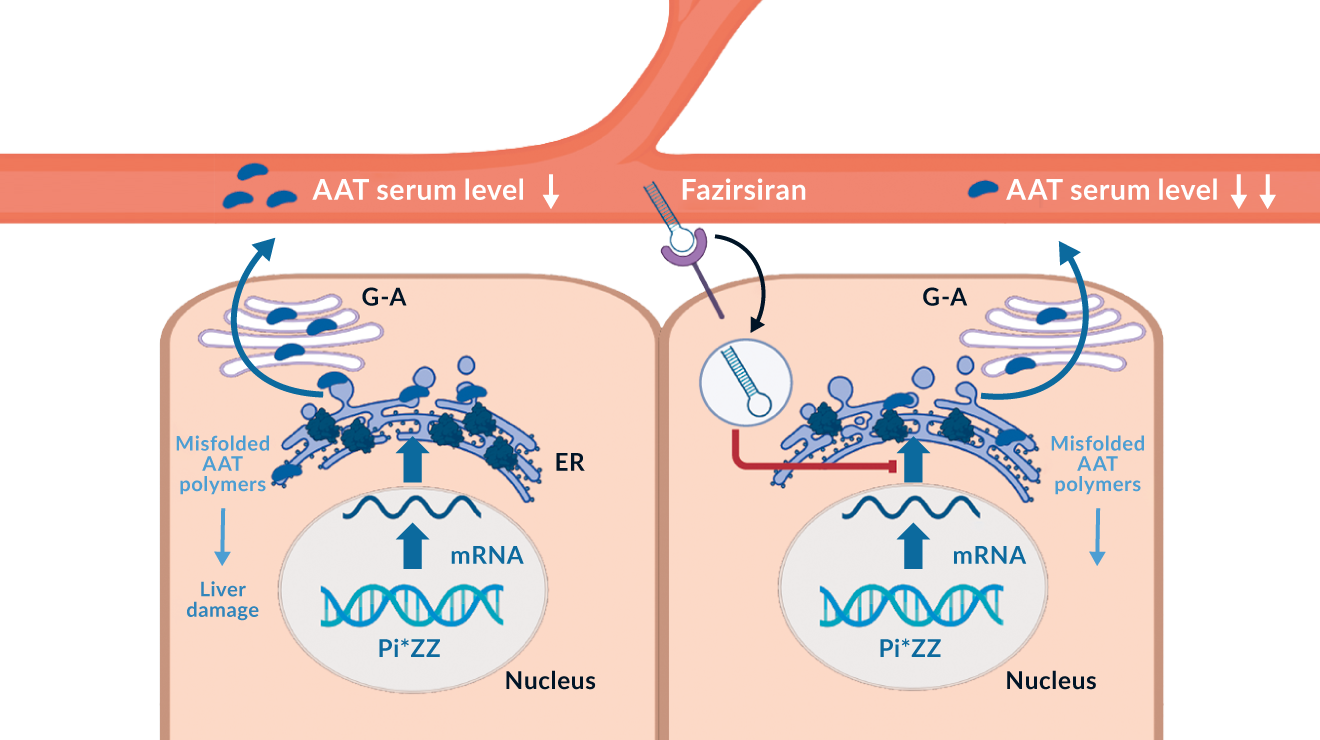 Figure 3. Biological effect of alpha-1 antitrypsin (AAT) mutation and the mode of action of fazirsiran, a small interfering RNA blocking AAT production
Figure 3. Biological effect of alpha-1 antitrypsin (AAT) mutation and the mode of action of fazirsiran, a small interfering RNA blocking AAT production
Key messages
- Alpha1-antitrypsin (AAT) is a protease inhibitor primarily produced in hepatocytes.
- Mutations in the SERPINA1-gene, encoding AAT, impair AAT production or secretion and result in alpha-1 antitrypsin deficiency (AATD).
- AATD predisposes to liver diseases caused by a proteotoxic “gain-of-function” and lung emphysema/chronic obstructive pulmonary disease caused by a “loss-of-function”.
- Over 100 different SERPINA1 mutations are known. The hetero- and homozygous Pi*Z variants (Pi*MZ/Pi*ZZ) constitute the most clinically relevant genotypes.
- Pi*ZZ is the predominant cause of severe AATD.
- Pi*ZZ patients can display prolonged jaundice as newborns or progressive liver fibrosis as adults.
- Pi*MZ patients typically require a second hit such as cystic fibrosis or non-alcoholic/alcoholic liver disease to develop significant liver fibrosis.
- In Pi*ZZ patients, weekly intravenous AAT substitution slows down the progression of lung disease.
- No specific treatment for AATD-associated liver disease exists, but AAT silencing with small interfering RNA showed promising results in a phase II clinical trial.
Future directions
In recent years, our knowledge about AATD-associated liver disease has expanded substantially. Despite this, a large number of people remain undiagnosed and their liver disease is misclassified. Many are recognised only at a stage of decompensated liver cirrhosis that is associated with a very poor prognosis and liver transplantation constitutes the only option. An proactive testing for AATD is needed to allow an early diagnosis. The latter should promote an adjustment of life-style, avoidance of risk factors and a family counseling. Patients with significant lung involvement should be offered an augmentation treatment as well as participation in promising clinical trials. Longitudinal studies are needed to better understand disease development as well as risk of liver cancer.
References
Abul-Husn NS, Cheng X, Li AH, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med. 2018;378: 1096-106.
Adam R, Karam V, Delvart V, et al. Evolution of indications and results of liver transplantation in europe. A report from the European Liver Transplant Registry (ELTR). J Hepatol. 2012;57: 675-88.
Alam H, Sehgal L, Kundu ST, Dalal SN, and Vaidya MM. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol Biol Cell. 2011;22: 4068-78.
Banga A, Gildea T, Rajeswaran J, Rokadia H, Blackstone EH, and Stoller JK. The natural history of lung function after lung transplantation for α(1)-antitrypsin deficiency. Am J Respir Crit Care Med, 2014;190: 274-81.
Blanco I, Bueno P, Diego I, et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis. 2017;12: 561-69.
Blanco I, Lipsker D, Lara B, and Janciauskiene S. Neutrophilic panniculitis associated with alpha-1-antitrypsin deficiency: an update. Br J Dermatol. 2016;174: 753-62.
Bornhorst JA, Greene DN, Ashwood ER, and Grenache DG. α1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013;143: 1000-08.
Cacciottolo TM, Gelson WT, Maguire G, Davies SE, and Griffiths WJ. Pi*Z heterozygous alpha-1 antitrypsin states accelerate parenchymal but not biliary cirrhosis. Eur J Gastroenterol Hepatol. 2014;26: 412-7.
Carey EJ, Iyer VN, Nelson DR, Nguyen JH, and Krowka MJ. Outcomes for recipients of liver transplantation for alpha-1-antitrypsin deficiency–related cirrhosis. Liver Transpl. 2013;19: 1370-6.
Cazzola M, Stolz D, Rogliani P, and Matera MG. α(1)-Antitrypsin deficiency and chronic respiratory disorders. Eur Respir Rev.2020;29.
Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe α1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;386: 360-8.
Christianson JC, Shaler TA, Tyler RE, and Kopito RR. OS-9 and GRP94 deliver mutant alpha1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat Cell Biol. 2008;10: 272-82.
Clark VC. Liver transplantation in alpha-1 antitrypsin deficiency. Clin Liver Dis. 2017;21: 355-65.
Clark VC, Marek G, Liu C, et al. Clinical and histologic features of adults with alpha-1 antitrypsin deficiency in a non-cirrhotic cohort. J Hepatol. 2018;69: 1357-64.
Elsensohn AN, Curtis JA, Secrest AM, et al. Alpha-1-antitrypsin deficiency panniculitis presenting with severe anasarca, pulmonary embolus and hypogammaglobulinaemia. Br J Dermatol. 2015;173: 289-91.
Feldman A, and Sokol RJ. Alpha-1-antitrypsin deficiency: an important cause of paediatric liver disease. Lung Health Prof Mag. 2013;4: 8-11.
Ferkingstad E, Oddsson A, Gretarsdottir S, et al. Genome-wide association meta-analysis yields 20 loci associated with gallstone disease. Nat Commun. 2018;9: 5101.
Ferrarotti I, Carroll TP, Ottaviani S, et al. Identification and characterisation of eight novel SERPINA1 Null mutations. Orphanet Journal of Rare Diseases. 2014;9: 172.
Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax. 2012;67: 669-74.
Foreman MG, Wilson C, DeMeo DL, et al. Alpha-1 Antitrypsin PiMZ genotype Is associated with chronic obstructive pulmonary disease in two racial groups. Ann Am Thorac Soc. 2017;14: 1280-87.
Franciosi AN, Ralph J, O'Farrell NJ, et al. Alpha-1 antitrypsin deficiency-associated panniculitis. J Am Acad Dermatol. 2022;87: 825-32.
Franciosiz AN, McCarthy C, Carroll TP, and McElvaney NG. Unusual acute sequelae of α1-antitrypsin deficiency: a myriad of symptoms with one common cure. Chest. 2015;148: e136-e38.
Fromme M, Oliverius M, and Strnad P. DEFI-ALFA: The French key to the alpha1 mystery?. Liver Int. 2019;39: 1019-21.
Fromme M, Schneider CV, Pereira V, et al. Hepatobiliary phenotypes of adults with alpha-1 antitrypsin deficiency. Gut. 2022a;71: 415-23.
Fromme M, Schneider CV, Trautwein C, Brunetti-Pierri N, and Strnad P. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J Hepatol. 2022b;76: 946-58.
Fu DA, and Campbell-Thompson M. Immunohistochemistry staining for human alpha-1 antitrypsin. Methods Mol Biol. 2017;1639: 139-43.
Greene CM, Marciniak SJ, Teckman J, et al. α1-Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2: 16051.
Guldiken N, Hamesch K, Schuller SM, et al. Mild iron overload as seen in individuals homozygous for the alpha-1 antitrypsin Pi*Z variant does not promote liver fibrogenesis in HFE knockout mice. Cells. 2019;8.
Hamesch K, Mandorfer M, Pereira VM, et al. Liver fibrosis and metabolic alterations in adults with alpha-1-antitrypsin deficiency caused by the Pi*ZZ mutation. Gastroenterology. 2019;157: 705-19.e18.
Hiller AM, Ekström M, Piitulainen E, Lindberg A, Rönmark E, and Tanash H. Cancer risk in severe alpha-1-antitrypsin deficiency. Eur Respir J. 2022;60.
Iskender I, Sakamoto J, Nakajima D, et al. Human α1-antitrypsin improves early posttransplant lung function: Pre-clinical studies in a pig lung transplant model. J Heart Lung Transplant. 2016;35: 913-21.
Janciauskiene S, Wrenger S, Immenschuh S, et al. The multifaceted effects of alpha1-antitrypsin on neutrophil functions. Frontiers in Pharmacology. 2018;9.
Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Köhnlein T, and Welte T. The discovery of α1-antitrypsin and its role in health and disease. Respir Med. 2011;105: 1129-39.
Kim RG, Nguyen P, Bettencourt R, et al. Magnetic resonance elastography identifies fibrosis in adults with alpha-1 antitrypsin deficiency liver disease: a prospective study. Aliment Pharmacol Ther. 2016;44: 287-99.
Kuhn BT, Veale A, Farah H, et al. Recombinant human alpha-1 antitrypsin (AAT) protein INBRX-101 demonstrates potential to achieve lung penetration and normal functional serum AAT levels in patients with AAT deficiency. American Journal of Respiratory and Critical Care Medicine. 2023;207: A4492.
Kuscuoglu D, Bewersdorf L, Wenzel K, et al. Dual proteotoxic stress accelerates liver injury via activation of p62-Nrf2. J Pathol. 2021;254: 80-91.
Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, and Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69: 927-47.
Lomas DA, Evans DL, Finch JT, and Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357: 605-7.
Lykavieris P, Ducot B, Lachaux A, et al. Liver disease associated with ZZ alpha1-antitrypsin deficiency and ursodeoxycholic acid therapy in children. J Pediatr Gastroenterol Nutr. 2008;47: 623-9.
Mahadeva R, Atkinson C, Li Z, et al. Polymers of Z alpha1-antitrypsin co-localise with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. 2005;166: 377-86.
McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of α1 proteinase inhibitor treatment for emphysema caused by severe α1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017;5: 51-60.
Merkel PA, Xie G, Monach PA, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol. 2017;69: 1054-66.
Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J. 2017;50.
Molloy K, Hersh CP, Morris VB, et al. Clarification of the risk of chronic obstructive pulmonary disease in α1-antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014;189: 419-27.
Mostafavi B, Piitulainen E, and Tanash HA. Survival in the Swedish cohort with alpha-1-antitrypsin deficiency, up to the age of 43-45 years. Int J Chron Obstruct Pulmon Dis. 2019;14: 525-30.
Petersen MC, and Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98: 2133-223.
Piitulainen E, and Sveger T. Respiratory symptoms and lung function in young adults with severe alpha(1)-antitrypsin deficiency (PiZZ). Thorax. 2002;57: 705-8.
Picker N, Hagiwara M, Baumann S, et all. Liver disease epidemiology and burden in patients with alterations in plasma protein metabolism: German retrospective insurance claims analysis. World J Hepatol. 2023;15(10): 1127-1139
Rahmattulla C, Mooyaart AL, van Hooven D, et al. Genetic variants in ANCA-associated vasculitis: a meta-analysis. Ann Rheum Dis. 2016;75: 1687-92.
Regev A, Guaqueta C, Molina EG, et al. Does the heterozygous state of alpha-1 antitrypsin deficiency have a role in chronic liver diseases? Interim results of a large case-control study. J Pediatr Gastroenterol Nutr. 2006;43 Suppl 1: S30-5.
Ruiz M, Lacaille F, Berthiller J, et al. Liver disease related to alpha1-antitrypsin deficiency in French children: The DEFI-ALPHA cohort. Liver Int. 2019;39: 1136-46.
Ruiz M, Lacaille F, Schrader C, et al. Paediatric and adult liver disease in alpha-1 antitrypsin deficiency. Semin Liver Dis. 2023;43: 258-66.
Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016;3: 668-82.
Schneider CV, Hamesch K, Gross A, et al. Liver phenotypes of European adults heterozygous or homozygous for Pi∗Z variant of AAT (Pi∗MZ vs Pi∗ZZ genotype) and noncarriers. Gastroenterology. 2020;159: 534-48.e11.
Stender S, Kozlitina J, Nordestgaard BG, Tybjærg-Hansen A, Hobbs HH, and Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet. 2017;49: 842-47.
Stolk J, Tov N, Chapman KR, et al. Efficacy and safety of inhaled α1-antitrypsin in patients with severe α1-antitrypsin deficiency and frequent exacerbations of COPD. Eur Respir J. 2019;54.
Stockley RA, Titlestad I, Tanash H, et al. Alvelestat, an oral neutrophil elastase inhibitor in alpha-1 antitrypsin deficiency (AATD): result of a phase II trial. American Journal of Respiratory and Critical Care Medicine. 2023;207: A4493.
Strange C, Moseley MA, Jones Y, Schwarz L, Xie L, and Brantly ML. Genetic testing of minors for alpha1-antitrypsin deficiency. Arch Pediatr Adolesc Med. 2006;160: 531-4.
Strnad P, Buch S, Hamesch K, et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut. 2019;68: 1099-107.
Strnad P, McElvaney NG, and Lomas DA. Alpha(1)-Antitrypsin Deficiency. N Engl J Med. 2020;382: 1443-55.
Strnad P, Nuraldeen R, Guldiken N, et al. Broad spectrum of hepatocyte inclusions in humans, animals, and experimental models. Compr Physiol. 2013;3: 1393-436.
Strnad P, and San Martin J. RNAi therapeutics for diseases involving protein aggregation: fazirsiran for alpha-1 antitrypsin deficiency-associated liver disease. Expert Opin Investig Drugs. 2023;32: 571-81.
Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200, 000 infants. N Engl J Med. 1976;294: 1316-21.
Sveger T, and Eriksson S. The liver in adolescents with alpha 1-antitrypsin deficiency. Hepatology. 1995;22: 514-7.
Tanash HA, Ekström M, Rönmark E, Lindberg A, and Piitulainen E. Survival in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison to a general population with known smoking habits. Eur Respir J. 2017;50.
Tanash HA, Riise GC, Hansson L, Nilsson PM, and Piitulainen E. Survival benefit of lung transplantation in individuals with severe α1-anti-trypsin deficiency (PiZZ) and emphysema. J Heart Lung Transplant. 2011;30: 1342-7.
Teckman JH. Liver disease in alpha-1 antitrypsin deficiency: current understanding and future therapy. Copd. 2013;10 Suppl 1: 35-43.
Teckman JH, and Blomenkamp KS. Pathophysiology of alpha-1 antitrypsin deficiency liver disease. Methods Mol Biol. 2017;1639: 1-8.
Teckman JH, and Perlmutter DH. Retention of mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is associated with an autophagic response. Am J Physiol Gastrointest Liver Physiol. 2000;279: G961-74.
Wedzicha JAEC-C, Miravitlles M, Hurst JR, et al. Management of COPD exacerbations: a European Respiratory Society/American Thoracic Society guideline. Eur Respir J. 2017;49.
Williams R, Aspinall R, Bellis M, et al. Addressing liver disease in the UK: a blueprint for attaining excellence in health care and reducing premature mortality from lifestyle issues of excess consumption of alcohol, obesity, and viral hepatitis. Lancet. 2014;384: 1953-97.