
12. Primary sclerosing cholangitis
Tobias J. Weismüller
Introduction
Primary sclerosing cholangitis (PSC) is a chronic progressive autoimmune cholestatic liver disease characterised by fibrotic strictures of the intra- and/or extrahepatic bile ducts. This causes the pathognomonic image of pearl cord-like bile duct dilatations in the cholangiogram. In a relevant number of patients, but not in all, biliary liver fibrosis and eventually cirrhosis occur as the disease progresses (Karlsen 2017, Weismuller 2017).
The disease is typically diagnosed in young patients (median age at diagnosis: approximately 40 years) and about 2/3 of patients are male. The epidemiology of PSC varies significantly between different geographic regions with highest incidences seen in Northern Europe and North America (Trivedi 2022). Incidence of PSC appears to be increasing according to epidemiological studies from the Netherlands and Sweden (Boonstra 2013, Lindkvist 2010) and is in these areas approximately 1/100000/year while the prevalence ranges around 10 / 100 000. There is a clear association with inflammatory bowel disease (IBD). In Northern Europe and North America, the prevalence of IBD in patients with PSC is 60–80%, with ulcerative colitis (CU) as the dominating subtype in more than 80% (Tsaitas, Semertzidou, and Sinakos 2014).
Diagnosis of PSC
PSC should be suspected in individuals with clinical and/or biochemical markers of cholestasis, especially when IBD is present; a detailed cholangiogram showing the characteristic biliary tract changes confirms the diagnosis, when secondary causes of sclerosing cholangitis and other biliary diseases have been excluded (Figure 1).
Symptoms at initial diagnosis of PSC include nonspecific general symptoms such as fatigue, lassitude, right upper abdominal pain, or weight loss, as well as clinical signs of cholestasis such as jaundice and pruritus, which can be accompanied by fever and chills in case of acute bacterial cholangitis. However, more than one third of patients are asymptomatic at initial diagnosis and are only incidentally diagnosed with elevated cholestasis parameters during routine laboratory checks. In particular, alkaline phosphatase (AP) is elevated three- to tenfold in 90% of patients, whereas aminotransferases are not or only minimally elevated. Serum bilirubin can be seen as a marker of late-stage disease, which increases when the fibrotic inflammation of the biliary tract leads to high grade strictures or to severe ductopenia (“pruning”). Autoantibodies do not play a role to confirm the diagnosis of PSC itself but to rule out variant syndromes or other autoimmune or cholestatic liver diseases. Physical examination at initial diagnosis rarely shows hepatomegaly or splenomegaly (Broome 1996, Tischendorf 2007) but results in normal findings in most patients. Abdominal ultrasound also frequently shows normal findings in early PSC, but sometimes liver parenchymal changes, cholestasis, or hilar lymphadenopathy can already be apparent.
 Figure 1. Differential diagnosis of PSC: Diseases of the liver and those affecting the liver, which can lead to features of sclerosing cholangitis. The differential diagnostic considerations in visually apparent sclerosing cholangitis cover a diverse array of conditions apart from PSC.
Figure 1. Differential diagnosis of PSC: Diseases of the liver and those affecting the liver, which can lead to features of sclerosing cholangitis. The differential diagnostic considerations in visually apparent sclerosing cholangitis cover a diverse array of conditions apart from PSC.
 Figure 2. Cholangiogram in PSC: ERC (left image) or MRI / MRCP (right image).
Figure 2. Cholangiogram in PSC: ERC (left image) or MRI / MRCP (right image).
According to current guidelines (European Association for the Study of the Liver 2022, Bowlus 2023) detailed imaging of the extra- and intrahepatic bile ducts by magnetic resonance cholangiography (MRC) is the crucial step in the diagnosis of PSC. The cholangiogram shows the characteristic bile duct changes with multifocal segmental strictures and consecutive dilatations of the intra- and/or extrahepatic bile ducts (Figure 2). Due to the much higher risk potential (especially iatrogenic pancreatitis and cholangitis) endoscopic retrograde cholangiography (ERC) is limited to cases when a therapeutic or diagnostic biliary intervention is indicated. Furthermore, MRI offers the advantage of imaging the hepatic parenchyma and visualising dilated bile duct segments proximal to complete stenoses.
Only in patients in early stages or in rare cases when image quality is limited by metallic implants or ascites, diagnostic ERC can be performed after weighing the risks and the expected therapeutic consequences.
Liver biopsy should be performed when high-quality MRI with MRCP is normal but elevated serum markers of cholestasis, especially in patients with IBD, raise the suspicion of small-duct PSC. Typical histopathological findings include “onion-skin”-like periductal fibrosis, fibroobliterative cholangitis, ductular reaction or ductopenia, periductal and sometimes also portal inflammation. Liver biopsy can also be considered in patients with markedly elevated transaminases, high IgG levels, and positive autoantibodies to corroborate the diagnosis of the variant syndrome of PSC with features of autoimmune hepatitis (AIH) (Boberg 2011).
Figure 3 summarises the diagnostic algorithm, when PSC is suspected.
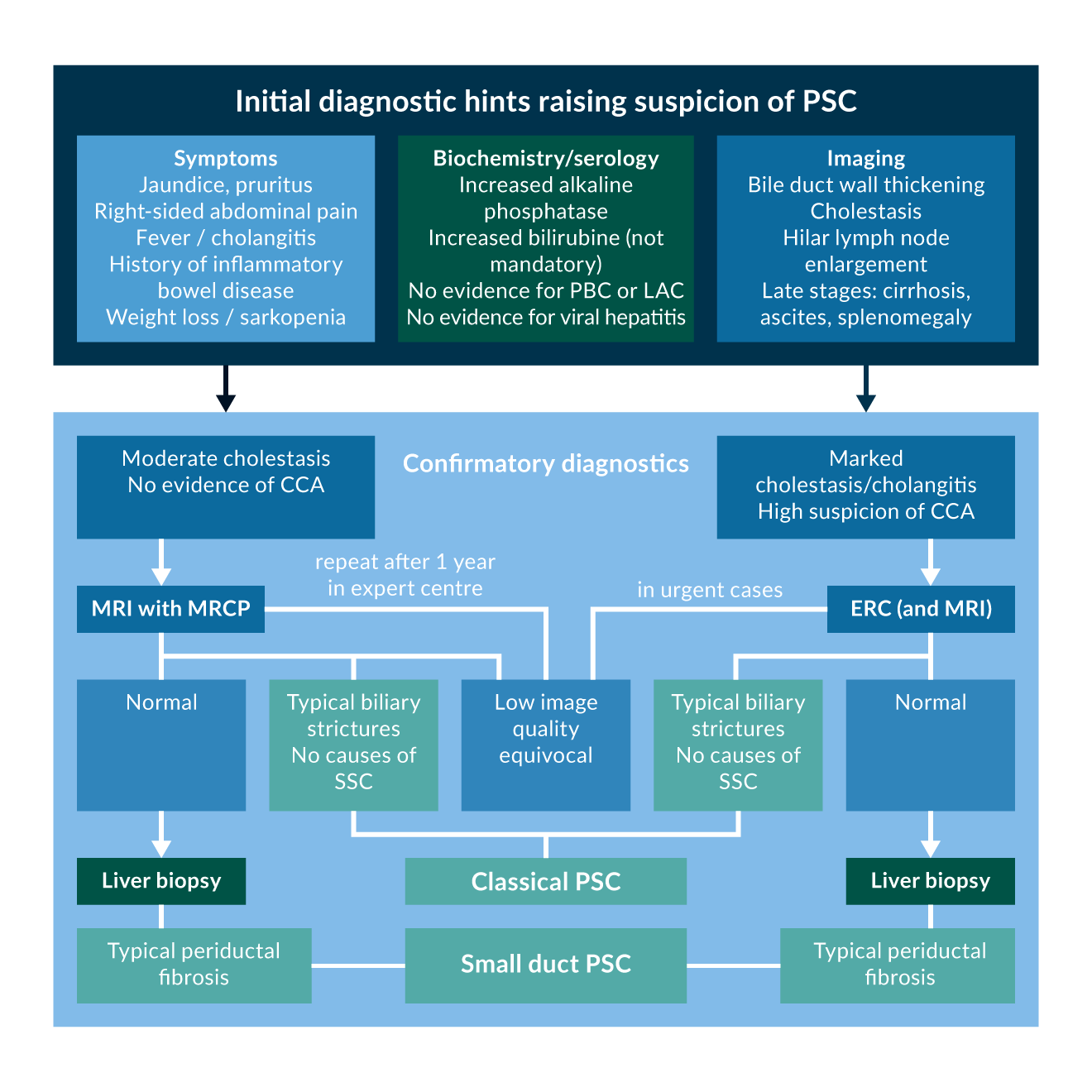 Figure 3. Diagnostic algorithm for PSC.
Figure 3. Diagnostic algorithm for PSC.Abbreviations: PSC: Primary Sclerosing Cholangitis, PBC: Primary Biliary Cholangitis, IAC: IgG4 associated cholangitis, CCA: Cholangiocarcinoma, MRI: Magnetic Resonance Imaging, MRCP: Magnetic Resonance Cholangiopancreaticography, ERC: Endoscopic Retrograde Cholangiography
Variant Syndromes and secondary causes of sclerosing cholangitis
Small-duct PSC (sdPSC) is a variant subtype which is diagnosed in about 4% of all PSC patients, when typical biochemical and histological features of PSC are present while a high-quality cholangiogram shows normal bile ducts (Kaplan 2007, Weismuller 2017, Bjornsson 2008). Other cholangiopathies such as PBC, ABCB4 deficiency, sarcoidosis, eosinophilic or IgG4-associated cholangitis must be excluded serologically and/or histologically. As compared with classical “large-duct” PSC this variant has a more benign disease course with only a minority of patients developing large-duct involvement and with a much lower risk of progression to end-stage liver disease or to cholangiocarcinoma.
PSC with features of AIH or so-called PSC-AIH-Overlap is another variant subtype that can be assumed when patients with a diagnosis of PSC show biochemical, serological, and/or histological features that overlap with those of AIH before or at the time of diagnosis of PSC, or later during the disease. Whether this is an independent syndrome or merely the coexistence of two separate diseases is still controversial. Applying the revised IAIHG scoring system 6–9% of PSC patients meet the diagnostic criteria of at least probable AIH (Boberg 2011, Weismuller 2017) and in paediatric PSC the frequency of features of AIH is even higher than 30% (Deneau 2017). According to the European Association for the Study of the Liver (EASL) clinical practice guidelines (European Association for the Study of the Liver 2022) a liver biopsy should be considered in PSC patients with alanine aminotransferase (ALT) levels five times the upper limit of normal (>5x ULN) and/or serum IgG levels > 1.5x ULN. If histology shows moderate to severe interface hepatitis in a patient with established PSC the co-existence or overlap with AIH can be diagnosed. Although there is not much systematic evidence regarding the course of the PSC-AIH-variant and its treatment options, management of the AIH component is based on the recommendations for the management of classical AIH.
IgG4-associated cholangitis (IAC) is the biliary phenotype of IgG4 related diseases (IgG4-RD), a systemic disease characterised by involvement of different organs, especially exocrine glands like pancreas but also retroperitoneum and bile ducts. The diagnosis of IgG4-RD is based on serology with markedly elevated IgG4-levels (>4 times) and on histology, imaging, other organ involvement and response to therapy (HISORt criteria). The biliary phenotype impressively resembles that of PSC, both in cholangiography and in clinical presentation (Lohr 2022). Since IAC in contrast to PSC responds well to corticosteroids it is crucial to check IgG4 serum levels in every patient with a first diagnosis of PSC and to exclude involvement of other organs.
IAC must be distinguished from another subtype of PSC in which borderline elevated serum levels of IgG4 can be detected without the HISORt criteria for a diagnosis of IAC being met. Evidence is accumulating in recent years that this subgroup of PSC with elevated IgG4-values has a significantly inferior transplant-free survival (Mendes 2006, Zhou 2021). It is not yet clear whether this subgroup would also benefit from anti-inflammatory treatment with corticosteroids.
There is a large number of different aetiologies of bile duct damage, which all share morphologic features of sclerosing cholangitis and should always be considered before the diagnosis of PSC can be made. Secondary sclerosing cholangitis can be caused by ischemic or traumatic bile duct injuries, as well as different immunologic or infectious diseases, which are summarised in Figure 1.
Clinical course of PSC and risk assessment
The clinical course of PSC is generally characterised by an increased frequency of episodic cholestatic symptoms such as jaundice, pruritus, and right-sided upper abdominal pain. Moreover, inflammatory-fibrotic strictures of the large and middle-sized bile ducts favour the development of ascending cholangitis up to severe cholangiosepsis. In advanced disease stage – but not in every patient – chronic cholangitis leads to biliary hepatic fibrosis and liver cirrhosis with complications of portal hypertension such as oesophageal varices and ascites. The overall disease course in PSC is highly variable, with some patients requiring transplantation shortly after diagnosis, while others live almost without symptom for decades. Depending on whether data from transplant centres (Broome 2002, Tischendorf 2007) or from large epidemiologic studies are analysed (Boonstra 2013), the median transplant-free survival after initial diagnosis is estimated between 10 and 20 years. Only about one third of patients die of liver failure, whereas carcinoma of the biliary tract or colon is the cause of death in more than 40% of patients (Bergquist 2002, Boonstra 2013, Claessen 2009).
Assessment of individual prognosis is difficult in a disease like PSC with a highly variable and often relapsing course. Several cohort studies identified the following clinical parameters as predictors of an unfavourable course with reduced transplant-free survival: older age at first diagnosis, coexisting ulcerative colitis, coexisting extrahepatic and extraintestinal autoimmune disease, splenomegaly, hepatomegaly, variceal hemorrhage, high-grade (“dominant”) strictures in the cholangiogram, extent of cholangiographic bile duct changes (Amsterdam score), liver elastography and extent of histologic changes (De Vries 2017, Kim 2000, Ponsioen 2010, Rupp, Tischendorf 2007, Corpechot 2014, Eaton 2016). In addition, the following biomarkers have been shown to be useful for prognostic assessment: Alkaline phosphatase (AP), bilirubin, aspartate aminotransferase (AST), and serum albumine (de Vries, Beuers, and Ponsioen 2015). Several prognostic scores for estimating prognosis in PSC have been developed: The revised Mayo-Risk-Score (Kim 2000) was the first risk score and calculates survival probability at 1 and 4 years based on patient age, bilirubine, albumine, AST and history of variceal bleeding. The Amsterdam-Oxford-Model (de Vries 2018) has also been validated in independent cohorts and calculates long-term overall survival at 5-, 10- and 15 years based on bilirubine, albumine, AST, alkaline phosphatase, platelets, large-duct-involvement and age at first diagnosis.
PSC and IBD
There is a clear association of PSC with IBD, and especially ulcerative colitis as the predominating subtype. IBD associated with PSC has typical features, particularly right-sided colitis with frequent "backwash ileitis" and rectal sparring (Loftus 2005). Conversely, PSC was found in 0.75% to 5.4% of patients with ulcerative colitis and in 1.2% to 3.4% of patients with Crohn's disease (Gizard 2014). Usually, IBD is diagnosed first and PSC later in the course of the disease or both diseases are diagnosed almost simultaneously; but sometimes IBD can manifest even years after the initial diagnosis of PSC. In order not to miss a clinically inapparent IBD, it is recommended that a complete ileocolonoscopy with random stepwise biopsies is always performed following the initial diagnosis of PSC.
The pathophysiological cause of this striking association of PSC and IBD is thought to be an aberrant homing of lymphocytes activated in the intestine, which migrate via the endothelium of the liver sinusoids into the liver and are involved in the establishment of a chronic hepatobiliary inflammatory process (Adams and Eksteen 2006).
Nevertheless, activity of both diseases does not show a regular pattern and it cannot be postulated that high activity of one disease leads to higher (or lower) activity of the other one. Furthermore, effective treatment of IBD does not have an impact on the course of PSC, even not with the α4β7-integrin-antagonists vedolizumab (Lynch 2020). Therefore, treatment strategies for IBD associated with PSC do not differ from those for IBD without PSC, with one exception: The risk of colorectal carcinoma in IBD patients is about 5 times higher and the carcinomas occur earlier in the course of the disease and are more frequently localised on the right side. Hence, international guidelines recommend yearly surveillance colonoscopies in all patients with IBD, as soon as additional PSC has been diagnosed (Magro 2017).
PSC as a risk factor for hepatobiliary malignancies
Even more than the risk of colorectal cancer, the risk of hepatobiliary malignancies is dramatically increased in PSC. According to population-based studies (de Valle, Bjornsson, and Lindkvist 2012, Boonstra 2013), the cumulative 10-year risk of cholangiocarcinoma is 6%-11%. In the very large cohort of the International PSC Study Group, recruiting mainly from tertiary centres, the annual incidence rate of hepatobiliary malignancies was 1.4/100 Pat. years. The risk for hepatobiliary malignancies was even higher in male patients, in patients with ulcerative colitis (compared to no IBD or Crohn’s disease) and in patients with classical PSC (compared to the small-duct variant or the PSC/AIH-variant) (Weismuller 2017). In addition to the more than hundred-fold increased risk of cholangiocarcinoma of the bile ducts including the gallbladder, a Swedish study also found an increased risk of pancreatic cancer (Bergquist 2002). Hepatocellular carcinomas (HCC), on the other hand, occurs comparatively rarely in PSC even in a cirrhotic stage of the disease (Zenouzi 2014).
Role of surveillance strategies
Given the markedly increased risk of malignancy, surveillance strategies for the early detection of carcinomas with the aim of enabling curative therapy are an important aspect in the management of PSC (Figure 4). Annual Colonoscopies to screen for colorectal neoplasia in all PSC patients with IBD are established and reduce significantly the colorectal cancer associated mortality (Boonstra 2013).
For cholangiocarcinoma, however, current publications on surveillance seem to paint a contradictory picture. In a prospective multicentre study from Sweden annual imaging with MRI/MRCP followed by ERCP and cytology/histology was not able to detect malignancy early enough to improve long-term survival (Villard 2023). In another international retrospective study including 2975 PSC-patients from 28 centres, different surveillance strategies were compared. With regular scheduled imaging overall survival improved and patients diagnosed with hepatobiliary malignancy were more likely to be treated with potentially curative therapies (surgical resection or liver transplantation) (Bergquist 2023).
The diagnosis of cholangiocarcinoma in a curable stage (CC) remains a challenging task because imaging studies have a limited sensitivity due to the often intramural growth pattern of CCA in PSC. Therefore, CCA are visible as stenosis or tumour only in late stages precluding curative therapeutic approaches. Furthermore, stenoses upon cholangiography may be caused by inflammatory activity as well as neoplasia, so that also biochemical tests and intraductal biopsy procedures have a low sensitivity and specificity. Newer techniques with direct visualisation through single-operator cholangioscopy might be a promising approach.
Despite those difficulties in diagnosing CCA in PSC, international practice guidelines recommend yearly high quality MRI with MRCP and/or liver ultrasound (Figure 4). Furthermore, a clinical evaluation every 6-12 months and serum liver-related tests including bilirubin, ALP, AST, platelets, and PT are recommended. Carbohydrate antigen 19-9 (CA 19-9) is not suggested for surveillance but should be done in case of suspected CCA. When a high-grade stricture is suspicious for CCA, invasive evaluation with ERC and forceps biopsy or brush cytology should be performed. Fluorescence in situ hybridisation (FISH) or equivalent chromosomal assessments can be considered when brush cytology and/or histology are equivocal. Due to their low sensitivity strictures should be reevaluated after 3 months when malignancy or dysplasia was not confirmed.
 Figure 4. Surveillance algorithm for PSC
Figure 4. Surveillance algorithm for PSCAbbreviations: PSC: Primary Sclerosing Cholangitis, IBD: Inflammatory Bowel Disease, CCA: Cholangiocarcinoma, HCC: hepatocellular carcinoma, MRI: Magnetic Resonance Imaging, MRCP: Magnetic Resonance Cholangiopancreaticography, ERC: Endoscopic Retrograde Cholangiography, CT: Computer Tomography
Medical treatment
The pathogenesis of PSC is largely unknown and, accordingly, a causal medical therapy for the disease is not yet available. Only antibiotic therapy for acute infectious cholangitis and endoscopic interventional therapy for obstructive cholestasis are established (Weismuller and Lankisch 2011).
Ursodeoxycholic acid (UDCA), a bile acid with choleretic, antiapoptotic, and antiinflammatory properties, has been used for cholestatic diseases including PSC for more than three decades, although evidence for a significant improvement in transplant-free survival has (in contrast to PBC) not yet been demonstrated in any PSC study. With a very high dosage of 28-30 mg/kg BW, even an opposite effect including disease progression was observed (Lindor 2009). In the studies with moderate dosages of UDCA (17-23 mg/kg BW), there were no significant improvements in hard endpoints, but at least a reduction in cholestasis parameters, especially AP. Subsequent subgroup analyses demonstrated that patients who experienced a significant reduction in AP with UDCA had significantly better transplant-free survival (Lindstrom 2013). Therefore, it is hypothesised that UDCA therapy at a moderate dose could improve prognosis in patients with a good biochemical response.
Despite ample evidence of an immunologically mediated pathogenesis of the disease, several smaller, but methodologically questionable (nonrandomised, nonplacebo-controlled) studies have failed to demonstrate prognostic improvement in PSC with the use of immunosuppressive or immunomodulatory agents (Culver and Chapman). However, there are two subgroups or variants of patients with PSC in whom immunosuppression is required: first, patients with coexisting autoimmune hepatitis (AIH) should be treated with immunosuppression according to current recommendations. A German retrospective multicentre study showed that PSC patients receiving immunosuppressive treatment already had more frequent signs of cirrhosis and elevated serum immunoglobulin G at diagnosis; also, the simplified AIH score and the modified histologic activity index were significantly elevated at therapy initiation (Schulze 2015). On the other hand, patients with IgG4-associated cholangitis (IAC) also benefit from immunosuppressive therapy and then show rapid regression of strictures on cholangiogram; however, it is unclear whether patients with PSC and only moderately elevated serum IgG4 also benefit from immunosuppression.
Antibiotics are an indispensable part of the therapy of acute bacterial cholangitis. In addition, the long-term use of various antibiotics such as metronidazole or vancomycin was evaluated in pilot studies and showed at least an improvement of biochemical, and in some cases also clinical and histological parameters (Davies 2008, Farkkila 2004, Tabibian 2014, Tabibian 2013). An adaptation of the antibiotic therapy strategy to the respective biliary germ spectrum seems to be reasonable at least in case of acute cholangitis (Negm 2010).
The management of cholestatic pruritus in PSC includes endoscopic treatment of relevant bile duct strictures and general measures such as moisturising creams and cooling. As medical first-line treatment option bezafibrate (400 mg daily) should be considered, taking into account relevant side effects (myalgias, myopathies, kidney dysfunction or worsening of liver parameters). In a randomised, placebo-controlled study, bezafibrate led to a reduction in pruritus of more than 40% within 3 weeks in 45% of participants in a mixed PBC and PSC cohort (de Vries 2021). Rifampicin (150-300 mg/d) is an alternative treatment option, but should initially only be given under close monitoring of liver values due to the risk of drug-induced toxic hepatitis (up to 12% of patients after 4-12 weeks) (European Association for the Study of the Liver 2022).
Endoscopic treatment
Fibrotic strictures of the bile ducts with consecutive bile duct dilatations characterise PSC, promote ascending cholangitis, and lead to cholestasis. Mechanical balloon dilatation (Figure 5) of these strictures using ERC can effectively resolve cholestasis and relieve symptoms of cholestasis (Gluck 2008, Gotthardt 2010). According to a recently published international randomised trial (Ponsioen 2018), short-term stent insertion is not less effective than ballon dilatation but was associated with significantly more complications. Therefore, balloon dilatation should be the preferred method, and stent implantation of PSC-associated bile duct strictures should be used cautiously only in selected cases. Choledocholithiasis, which is occasionally associated with PSC, can also be treated endoscopically by means of balloon or basket assisted stone removal or mechanical or electrohydraulic lithotripsy.
Apart from the therapy of cholestasis, ERC also holds an important role in the diagnostic work-up of new biliary strictures suspicious for malignancy. For this purpose, imaging techniques such as cholangioscopy and intraductal ultrasound, as well as brush or forceps biopsy to obtain cells and tissue are common methods (Aabakken 2017). The European Association for the Study of the Liver recommends in its current guideline (European Association for the Study of the Liver 2022) performing an ERC in patients with relevant (symptomatic) strictures and in cases of suspected cholangiocarcinoma, but not for routine surveillance.
 Figure 5. Endoscopic treatment of a high-grade stricture (A) with either balloon dilatation (B) or stenting (C). Combination of both methods is also frequently applied.
Figure 5. Endoscopic treatment of a high-grade stricture (A) with either balloon dilatation (B) or stenting (C). Combination of both methods is also frequently applied.
Liver transplantation for PSC
In the absence of definitive drug or endoscopic treatment options, liver transplantation (LT) remains the only curative therapy for PSC. Currently between five and fifteen percent of liver transplants in Europe are performed for this indication, depending on the region (Adam 2018, Fosby 2015). Against the background of the variable disease course and the incalculable risk of malignancy, the greatest challenge for an optimal prognosis for PSC-patients is to ensure that a donor organ is available at the right time. Compared with other hepatopathies, PSC progresses rather slowly but can also worsen acutely in the setting of cholangiosepsis. On the other hand, preemptive liver transplantation carries a higher short-term mortality risk of LT itself than the short-term natural course of the disease. However, when cholangiocarcinoma (CCA) is detected during disease progression, which is expected in 10-15% of patients, this is often advanced and therefore a contraindication to transplantation.
PSC patients should therefore be listed for LT when life expectancy and quality of life are so limited due to portal hypertension, liver failure, recurrent cholangitis or refractory pruritus that transplantation represents the lower risk compared to spontaneous progression (Martin and Levy 2017). Of note, the urgency of liver transplantation is not adequately reflected by the MELD score, which was designed for cirrhotic patients but underrates the main mortality factors in PSC like pronounced cholestasis, frequent cholangitis, acute cholangiosepsis and CCA.
Hence, many transplant allocation systems established exceptional points for PSC-patients, when they fulfill certain criteria, which reflect urgency better than the MELD-Score alone. This allows higher priority for PSC patients on the waiting list and allows a large proportion of PSC patients to be provided with a donor organ in a timely manner and keeps waiting list mortality low.
Long-term survival of organ recipients, most of whom are relatively young for this indication, is comparatively good with 79% to 85% after 5 years and 70%-80% after 10 years (Hildebrand 2016, Ravikumar 2015). However, biliary strictures, including PSC recurrence, occur in up to 36% of recipients transplanted for PSC and significantly affect both graft and patient survival. In case of cholestatic biochemistry following LT, it is recommended to initiate appropriate diagnostic steps without delay (MRI, in severe cholestasis also ERC, or liver biopsy); the treatment strategies in case of recurrent PSC do not differ from PSC-management in the pretransplant situation.
Risk factors for PSC recurrence in retrospective analyses were donor and/or recipient age, recipient INR prior to LT, cold ischaemia time, rejection and immunosuppression with tacrolimus. However, while these parameters could not be confirmed in meta-analyses (Buchholz 2018), almost all analyses identified (active) chronic inflammatory bowel disease as the most important risk factor for PSC recurrence. Whether intensified IBD therapy or even a protective colectomy could prevent recurrence is still under debate. The role of "prophylactic" continued UDCA therapy after LT is also unclear. The type of biliary anastomosis does not influence the risk of recurrence, so that no general recommendation is given for the creation of a biliodigestive anastomosis.
Even after LT, the risk of IBD-associated colon cancer is significantly increased. Annual screening colonoscopies are therefore strongly recommended. If no IBD is present, screening colonoscopies should be performed at least every 5 years, due to the increased risk of malignancy after LT.
References
Aabakken, L., T. H. Karlsen, J. Albert, M. Arvanitakis, O. Chazouilleres, J. M. Dumonceau, M. Farkkila, P. Fickert, G. M. Hirschfield, A. Laghi, M. Marzioni, M. Fernandez, S. P. Pereira, J. Pohl, J. W. Poley, C. Y. Ponsioen, C. Schramm, F. Swahn, A. Tringali, and C. Hassan. 2017. 'Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline', Endoscopy, 49: 588-608.
Adam, R., V. Karam, V. Cailliez, O. Grady JG, D. Mirza, D. Cherqui, J. Klempnauer, M. Salizzoni, J. Pratschke, N. Jamieson, E. Hidalgo, A. Paul, R. L. Andujar, J. Lerut, L. Fisher, K. Boudjema, C. Fondevila, O. Soubrane, P. Bachellier, A. D. Pinna, G. Berlakovich, W. Bennet, M. Pinzani, P. Schemmer, K. Zieniewicz, C. J. Romero, P. De Simone, B. G. Ericzon, S. Schneeberger, S. J. Wigmore, J. F. Prous, M. Colledan, R. J. Porte, S. Yilmaz, D. Azoulay, J. Pirenne, P. D. Line, P. Trunecka, F. Navarro, A. V. Lopez, L. De Carlis, S. R. Pena, E. Kochs, C. Duvoux, centers all the other 126 contributing, Liver the European, and Association Intestine Transplant. 2018. '2018 Annual Report of the European Liver Transplant Registry (ELTR) - 50-year evolution of liver transplantation', Transpl Int, 31: 1293-317.
Adams, D. H., and B. Eksteen. 2006. 'Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease', Nat Rev Immunol, 6: 244-51.
Bergquist, A., A. Ekbom, R. Olsson, D. Kornfeldt, L. Loof, A. Danielsson, R. Hultcrantz, S. Lindgren, H. Prytz, H. Sandberg-Gertzen, S. Almer, F. Granath, and U. Broome. 2002. 'Hepatic and extrahepatic malignancies in primary sclerosing cholangitis', J Hepatol, 36: 321-7.
Bergquist, A., T. J. Weismuller, C. Levy, C. Rupp, D. Joshi, J. S. Nayagam, A. J. Montano-Loza, E. Lytvyak, E. Wunsch, P. Milkiewicz, R. Zenouzi, C. Schramm, N. Cazzagon, A. Floreani, I. F. Liby, M. Wiestler, H. Wedemeyer, T. Zhou, C. P. Strassburg, E. Rigopoulou, G. Dalekos, M. Narasimman, X. Verhelst, H. Degroote, M. Vesterhus, A. E. Kremer, B. Bundgens, F. Rorsman, E. Nilsson, K. K. Jorgensen, E. von Seth, M. Cornillet Jeannin, N. Nyhlin, H. Martin, S. Kechagias, K. Wiencke, M. Werner, B. T. Beretta-Piccoli, M. Marzioni, H. Isoniemi, J. Arola, A. Wefer, J. Soderling, M. Farkkila, H. Lenzen, and P. S. C. Study Group International. 2023. 'Impact on follow-up strategies in patients with primary sclerosing cholangitis', Liver Int, 43: 127-38.
Bjornsson, E., R. Olsson, A. Bergquist, S. Lindgren, B. Braden, R. W. Chapman, K. M. Boberg, and P. Angulo. 2008. 'The natural history of small-duct primary sclerosing cholangitis', Gastroenterology, 134: 975-80.
Boberg, K. M., R. W. Chapman, G. M. Hirschfield, A. W. Lohse, M. P. Manns, and E. Schrumpf. 2011. 'Overlap syndromes: the International Autoimmune Hepatitis Group (IAIHG) position statement on a controversial issue', J Hepatol, 54: 374-85.
Boonstra, K., R. K. Weersma, K. J. van Erpecum, E. A. Rauws, B. W. Spanier, A. C. Poen, K. M. van Nieuwkerk, J. P. Drenth, B. J. Witteman, H. A. Tuynman, A. H. Naber, P. J. Kingma, H. R. van Buuren, B. van Hoek, F. P. Vleggaar, N. van Geloven, U. Beuers, and C. Y. Ponsioen. 2013. 'Population-based epidemiology, malignancy risk, and outcome of primary sclerosing cholangitis', Hepatology, 58: 2045-55.
Bowlus, C. L., L. Arrive, A. Bergquist, M. Deneau, L. Forman, S. I. Ilyas, K. E. Lunsford, M. Martinez, G. Sapisochin, R. Shroff, J. H. Tabibian, and D. N. Assis. 2023. 'AASLD practice guidance on primary sclerosing cholangitis and cholangiocarcinoma', Hepatology, 77: 659-702.
Broome, U., H. Glaumann, E. Lindstom, L. Loof, S. Almer, H. Prytz, H. Sandberg-Gertzen, S. Lindgren, F. T. Fork, G. Jarnerot, and R. Olsson. 2002. 'Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC)', J Hepatol, 36: 586-9.
Broome, U., R. Olsson, L. Loof, G. Bodemar, R. Hultcrantz, A. Danielsson, H. Prytz, H. Sandberg-Gertzen, S. Wallerstedt, and G. Lindberg. 1996. 'Natural history and prognostic factors in 305 Swedish patients with primary sclerosing cholangitis', Gut, 38: 610-5.
Buchholz, B. M., P. M. Lykoudis, R. Ravikumar, J. M. Pollok, and G. K. Fusai. 2018. 'Role of colectomy in preventing recurrent primary sclerosing cholangitis in liver transplant recipients', World J Gastroenterol, 24: 3171-80.
Claessen, M. M., F. P. Vleggaar, K. M. Tytgat, P. D. Siersema, and H. R. van Buuren. 2009. 'High lifetime risk of cancer in primary sclerosing cholangitis', J Hepatol, 50: 158-64.
Corpechot, C., F. Gaouar, A. El Naggar, A. Kemgang, D. Wendum, R. Poupon, F. Carrat, and O. Chazouilleres. 2014. 'Baseline values and changes in liver stiffness measured by transient elastography are associated with severity of fibrosis and outcomes of patients with primary sclerosing cholangitis', Gastroenterology, 146: 970-9; quiz e15-6.
Culver, E. L., and R. W. Chapman. 'Systematic review: management options for primary sclerosing cholangitis and its variant forms - IgG4-associated cholangitis and overlap with autoimmune hepatitis', Aliment Pharmacol Ther, 33: 1273-91.
Davies, Y. K., K. M. Cox, B. A. Abdullah, A. Safta, A. B. Terry, and K. L. Cox. 2008. 'Long-term treatment of primary sclerosing cholangitis in children with oral vancomycin: an immunomodulating antibiotic', J Pediatr Gastroenterol Nutr, 47: 61-7.
de Valle, M. B., E. Bjornsson, and B. Lindkvist. 2012. 'Mortality and cancer risk related to primary sclerosing cholangitis in a Swedish population-based cohort', Liver Int, 32: 441-8.
de Vries, E., R. Bolier, J. Goet, A. Pares, J. Verbeek, M. de Vree, J. Drenth, K. van Erpecum, K. van Nieuwkerk, F. van der Heide, N. Mostafavi, J. Helder, C. Ponsioen, R. Oude Elferink, H. van Buuren, U. Beuers, and Group Netherlands Association for the Study of the Liver-Cholestasis Working. 2021. 'Fibrates for Itch (FITCH) in Fibrosing Cholangiopathies: A Double-Blind, Randomized, Placebo-Controlled Trial', Gastroenterology, 160: 734-43 e6.
de Vries, E. M., U. Beuers, and C. Y. Ponsioen. 2015. 'Biomarkers for disease progression of primary sclerosing cholangitis', Curr Opin Gastroenterol, 31: 239-46.
De Vries, E. M., M. de Krijger, M. Farkkila, J. Arola, P. Schirmacher, D. Gotthardt, B. Goeppert, P. J. Trivedi, G. M. Hirschfield, H. Ytting, B. Vainer, H. R. van Buuren, K. Biermann, M. H. Harms, O. Chazouilleres, D. Wendum, A. D. Kemgang, R. W. Chapman, L. M. Wang, K. D. Williamson, A. S. Gouw, V. Paradis, C. Sempoux, U. Beuers, S. Hubscher, J. Verheij, and C. Ponsioen. 2017. 'Validation of the Prognostic Value of Histologic Scoring Systems in Primary Sclerosing Cholangitis; An International Cohort Study', Hepatology.
de Vries, E. M., J. Wang, K. D. Williamson, M. M. Leeflang, K. Boonstra, R. K. Weersma, U. Beuers, R. W. Chapman, R. B. Geskus, and C. Y. Ponsioen. 2018. 'A novel prognostic model for transplant-free survival in primary sclerosing cholangitis', Gut, 67: 1864-69.
Deneau, M. R., W. El-Matary, P. L. Valentino, R. Abdou, K. Alqoaer, M. Amin, A. Z. Amir, M. Auth, F. Bazerbachi, A. Broderick, A. Chan, J. Cotter, S. Doan, M. El-Youssef, F. Ferrari, K. N. Furuya, M. Gottrand, F. Gottrand, N. Gupta, M. Homan, B. M. Kamath, K. M. Kim, K. L. Kolho, A. Konidari, B. Koot, R. Iorio, O. Ledder, C. Mack, M. Martinez, T. Miloh, P. Mohan, N. O'Cathain, A. Papadopoulou, A. Ricciuto, L. Saubermann, P. Sathya, E. Shteyer, V. Smolka, A. Tanaka, R. Varier, V. Venkat, B. Vitola, M. B. Vos, M. Woynarowski, J. Yap, and M. K. Jensen. 2017. 'The natural history of primary sclerosing cholangitis in 781 children: A multicenter, international collaboration', Hepatology, 66: 518-27.
Eaton, J. E., B. Dzyubak, S. K. Venkatesh, T. C. Smyrk, G. J. Gores, R. L. Ehman, N. F. LaRusso, A. A. Gossard, and K. N. Lazaridis. 2016. 'Performance of magnetic resonance elastography in primary sclerosing cholangitis', J Gastroenterol Hepatol, 31: 1184-90.
European Association for the Study of the Liver. 2022. 'EASL Clinical Practice Guidelines on sclerosing cholangitis', J Hepatol, 77: 761-806.
Farkkila, M., A. L. Karvonen, H. Nurmi, H. Nuutinen, M. Taavitsainen, P. Pikkarainen, and P. Karkkainen. 2004. 'Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: a randomized placebo-controlled trial', Hepatology, 40: 1379-86.
Fosby, B., E. Melum, K. Bjoro, W. Bennet, A. Rasmussen, I. M. Andersen, M. Castedal, M. Olausson, C. Wibeck, M. Gotlieb, H. Gjertsen, L. Toivonen, S. Foss, H. Makisalo, A. Nordin, T. Sanengen, A. Bergquist, M. E. Larsson, G. Soderdahl, G. Nowak, K. M. Boberg, H. Isoniemi, S. Keiding, A. Foss, P. D. Line, S. Friman, E. Schrumpf, B. G. Ericzon, K. Hockerstedt, and T. H. Karlsen. 2015. 'Liver transplantation in the Nordic countries - An intention to treat and post-transplant analysis from The Nordic Liver Transplant Registry 1982-2013', Scand J Gastroenterol, 50: 797-808.
Gizard, E., A. C. Ford, J. P. Bronowicki, and L. Peyrin-Biroulet. 2014. 'Systematic review: The epidemiology of the hepatobiliary manifestations in patients with inflammatory bowel disease', Aliment Pharmacol Ther, 40: 3-15.
Gluck, M., N. R. Cantone, J. J. Brandabur, D. J. Patterson, J. E. Bredfeldt, and R. A. Kozarek. 2008. 'A twenty-year experience with endoscopic therapy for symptomatic primary sclerosing cholangitis', J Clin Gastroenterol, 42: 1032-9.
Gotthardt, D. N., G. Rudolph, P. Kloters-Plachky, H. Kulaksiz, and A. Stiehl. 2010. 'Endoscopic dilation of dominant stenoses in primary sclerosing cholangitis: outcome after long-term treatment', Gastrointest Endosc, 71: 527-34.
Hildebrand, T., N. Pannicke, A. Dechene, D. N. Gotthardt, G. Kirchner, F. P. Reiter, M. Sterneck, K. Herzer, H. Lenzen, C. Rupp, H. Barg-Hock, P. de Leuw, A. Teufel, V. Zimmer, F. Lammert, C. Sarrazin, U. Spengler, C. Rust, M. P. Manns, C. P. Strassburg, C. Schramm, T. J. Weismuller, and P. S. C. Study Group German. 2016. 'Biliary strictures and recurrence after liver transplantation for primary sclerosing cholangitis: A retrospective multicenter analysis', Liver Transpl, 22: 42-52.
Kaplan, G. G., K. B. Laupland, D. Butzner, S. J. Urbanski, and S. S. Lee. 2007. 'The burden of large and small duct primary sclerosing cholangitis in adults and children: a population-based analysis', Am J Gastroenterol, 102: 1042-9.
Karlsen, T. H., T. Folseraas, D. Thorburn, and M. Vesterhus. 2017. 'Primary sclerosing cholangitis - a comprehensive review', J Hepatol, 67: 1298-323.
Kim, W. R., T. M. Therneau, R. H. Wiesner, J. J. Poterucha, J. T. Benson, M. Malinchoc, N. F. LaRusso, K. D. Lindor, and E. R. Dickson. 2000. 'A revised natural history model for primary sclerosing cholangitis', Mayo Clin Proc, 75: 688-94.
Lindkvist, B., M. Benito de Valle, B. Gullberg, and E. Bjornsson. 2010. 'Incidence and prevalence of primary sclerosing cholangitis in a defined adult population in Sweden', Hepatology, 52: 571-7.
Lindor, K. D., K. V. Kowdley, V. A. Luketic, M. E. Harrison, T. McCashland, A. S. Befeler, D. Harnois, R. Jorgensen, J. Petz, J. Keach, J. Mooney, C. Sargeant, J. Braaten, T. Bernard, D. King, E. Miceli, J. Schmoll, T. Hoskin, P. Thapa, and F. Enders. 2009. 'High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis', Hepatology, 50: 808-14.
Lindstrom, L., R. Hultcrantz, K. M. Boberg, I. Friis-Liby, and A. Bergquist. 2013. 'Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis', Clin Gastroenterol Hepatol, 11: 841-6.
Loftus, E. V., Jr., G. C. Harewood, C. G. Loftus, W. J. Tremaine, W. S. Harmsen, A. R. Zinsmeister, D. A. Jewell, and W. J. Sandborn. 2005. 'PSC-IBD: a unique form of inflammatory bowel disease associated with primary sclerosing cholangitis', Gut, 54: 91-6.
Lohr, J. M., M. Vujasinovic, J. Rosendahl, J. H. Stone, and U. Beuers. 2022. 'IgG4-related diseases of the digestive tract', Nat Rev Gastroenterol Hepatol, 19: 185-97.
Lynch, K. D., R. W. Chapman, S. Keshav, A. J. Montano-Loza, A. L. Mason, A. E. Kremer, M. Vetter, M. de Krijger, C. Y. Ponsioen, P. Trivedi, G. Hirschfield, C. Schramm, C. H. Liu, C. L. Bowlus, D. J. Estes, D. Pratt, C. Hedin, A. Bergquist, A. C. de Vries, C. J. van der Woude, L. Yu, D. N. Assis, J. Boyer, H. Ytting, E. Hallibasic, M. Trauner, H. U. Marschall, L. M. Daretti, M. Marzioni, K. K. Yimam, N. Perin, A. Floreani, B. T. Beretta-Piccoli, J. K. Rogers, Group International Primary Sclerosing Cholangitis Study, and C. Levy. 2020. 'Effects of Vedolizumab in Patients With Primary Sclerosing Cholangitis and Inflammatory Bowel Diseases', Clin Gastroenterol Hepatol, 18: 179-87 e6.
Magro, F., P. Gionchetti, R. Eliakim, S. Ardizzone, A. Armuzzi, M. Barreiro-de Acosta, J. Burisch, K. B. Gecse, A. L. Hart, P. Hindryckx, C. Langner, J. K. Limdi, G. Pellino, E. Zagorowicz, T. Raine, M. Harbord, F. Rieder, Crohn's European, and Organisation Colitis. 2017. 'Third European Evidence-based Consensus on Diagnosis and Management of Ulcerative Colitis. Part 1: Definitions, Diagnosis, Extra-intestinal Manifestations, Pregnancy, Cancer Surveillance, Surgery, and Ileo-anal Pouch Disorders', J Crohns Colitis, 11: 649-70.
Martin, E. F., and C. Levy. 2017. 'Timing, Management, and Outcomes of Liver Transplantation in Primary Sclerosing Cholangitis', Semin Liver Dis, 37: 305-13.
Mendes, F. D., R. Jorgensen, J. Keach, J. A. Katzmann, T. Smyrk, J. Donlinger, S. Chari, and K. D. Lindor. 2006. 'Elevated serum IgG4 concentration in patients with primary sclerosing cholangitis', Am J Gastroenterol, 101: 2070-5.
Negm, A. A., A. Schott, R. P. Vonberg, T. J. Weismueller, A. S. Schneider, S. Kubicka, C. P. Strassburg, M. P. Manns, S. Suerbaum, J. Wedemeyer, and T. O. Lankisch. 2010. 'Routine bile collection for microbiological analysis during cholangiography and its impact on the management of cholangitis', Gastrointest Endosc, 72: 284-91.
Ponsioen, C. Y., U. Arnelo, A. Bergquist, E. A. Rauws, V. Paulsen, P. Cantu, I. Parzanese, E. M. De Vries, K. N. van Munster, K. Said, O. Chazouilleres, B. Desaint, A. Kemgang, M. Farkkila, S. Van der Merwe, W. Van Steenbergen, H. U. Marschall, P. O. Stotzer, D. Thorburn, S. P. Pereira, and L. Aabakken. 2018. 'No Superiority of Stents vs Balloon Dilatation for Dominant Strictures in Patients With Primary Sclerosing Cholangitis', Gastroenterology, 155: 752-59 e5.
Ponsioen, C. Y., J. B. Reitsma, K. M. Boberg, L. Aabakken, E. A. Rauws, and E. Schrumpf. 2010. 'Validation of a cholangiographic prognostic model in primary sclerosing cholangitis', Endoscopy, 42: 742-7.
Ravikumar, R., E. Tsochatzis, S. Jose, M. Allison, A. Athale, F. Creamer, B. Gunson, V. Iyer, M. Madanur, D. Manas, A. Monaco, D. Mirza, N. Owen, K. Roberts, G. Sen, P. Srinivasan, S. Wigmore, G. Fusai, B. Fernando, and A. Burroughs. 2015. 'Risk factors for recurrent primary sclerosing cholangitis after liver transplantation', J Hepatol, 63: 1139-46.
Rupp, C., A. Mummelthei, P. Sauer, K. H. Weiss, P. Schirmacher, A. Stiehl, W. Stremmel, and D. N. Gotthardt. 'Non-IBD immunological diseases are a risk factor for reduced survival in PSC', Liver Int, 33: 86-93.
Schulze, K., T. J. Weismuller, M. Bubenheim, P. Huebener, R. Zenouzi, H. Lenzen, C. Rupp, D. Gotthardt, P. de Leuw, A. Teufel, V. Zimmer, F. P. Reiter, C. Rust, L. Tharun, A. Quaas, S. A. Weidemann, F. Lammert, C. Sarrazin, M. P. Manns, A. W. Lohse, C. Schramm, and P. S. C. Study Group German. 2015. 'Criteria Used in Clinical Practice to Guide Immunosuppressive Treatment in Patients with Primary Sclerosing Cholangitis', PLoS One, 10: e0140525.
Tabibian, J. H., A. Gossard, M. El-Youssef, J. E. Eaton, J. Petz, R. Jorgensen, F. B. Enders, A. Tabibian, and K. D. Lindor. 2014. 'Prospective Clinical Trial of Rifaximin Therapy for Patients With Primary Sclerosing Cholangitis', Am J Ther.
Tabibian, J. H., E. Weeding, R. A. Jorgensen, J. L. Petz, J. C. Keach, J. A. Talwalkar, and K. D. Lindor. 2013. 'Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis - a pilot study', Aliment Pharmacol Ther, 37: 604-12.
Tischendorf, J. J., H. Hecker, M. Kruger, M. P. Manns, and P. N. Meier. 2007. 'Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: A single center study', Am J Gastroenterol, 102: 107-14.
Trivedi, P. J., C. L. Bowlus, K. K. Yimam, H. Razavi, and C. Estes. 2022. 'Epidemiology, Natural History, and Outcomes of Primary Sclerosing Cholangitis: A Systematic Review of Population-based Studies', Clin Gastroenterol Hepatol, 20: 1687-700 e4.
Tsaitas, C., A. Semertzidou, and E. Sinakos. 2014. 'Update on inflammatory bowel disease in patients with primary sclerosing cholangitis', World J Hepatol, 6: 178-87.
Villard, C., I. Friis-Liby, F. Rorsman, K. Said, A. Warnqvist, M. Cornillet, S. Kechagias, N. Nyhlin, M. Werner, I. Janczewska, T. Hagstrom, E. Nilsson, and A. Bergquist. 2023. 'Prospective surveillance for cholangiocarcinoma in unselected individuals with primary sclerosing cholangitis', J Hepatol, 78: 604-13.
Weismuller, T. J., and T. O. Lankisch. 2011. 'Medical and endoscopic therapy of primary sclerosing cholangitis', Best Pract Res Clin Gastroenterol, 25: 741-52.
Weismuller, T. J., P. J. Trivedi, A. Bergquist, M. Imam, H. Lenzen, C. Y. Ponsioen, K. Holm, D. Gotthardt, M. A. Farkkila, H. U. Marschall, D. Thorburn, R. K. Weersma, J. Fevery, T. Mueller, O. Chazouilleres, K. Schulze, K. N. Lazaridis, S. Almer, S. P. Pereira, C. Levy, A. Mason, S. Naess, C. L. Bowlus, A. Floreani, E. Halilbasic, K. K. Yimam, P. Milkiewicz, U. Beuers, D. K. Huynh, A. Pares, C. N. Manser, G. N. Dalekos, B. Eksteen, P. Invernizzi, C. P. Berg, G. I. Kirchner, C. Sarrazin, V. Zimmer, L. Fabris, F. Braun, M. Marzioni, B. D. Juran, K. Said, C. Rupp, K. Jokelainen, M. Benito de Valle, F. Saffioti, A. Cheung, M. Trauner, C. Schramm, R. W. Chapman, T. H. Karlsen, E. Schrumpf, C. P. Strassburg, M. P. Manns, K. D. Lindor, G. M. Hirschfield, B. E. Hansen, K. M. Boberg, and P. S. C. Study Group International. 2017. 'Patient Age, Sex, and Inflammatory Bowel Disease Phenotype Associate With Course of Primary Sclerosing Cholangitis', Gastroenterology, 152: 1975-84 e8.
Zenouzi, R., T. J. Weismuller, P. Hubener, K. Schulze, M. Bubenheim, N. Pannicke, C. Weiler-Normann, H. Lenzen, M. P. Manns, A. W. Lohse, and C. Schramm. 2014. 'Low risk of hepatocellular carcinoma in patients with primary sclerosing cholangitis with cirrhosis', Clin Gastroenterol Hepatol, 12: 1733-8.
Zhou, T., H. Lenzen, L. Dold, B. Bundgens, H. Wedemeyer, M. P. Manns, M. A. Gonzalez-Carmona, C. P. Strassburg, and T. J. Weismuller. 2021. 'Primary sclerosing cholangitis with moderately elevated serum-IgG4 - characterization and outcome of a distinct variant phenotype', Liver Int, 41: 2924-33.